DeepCKI:一个基于变分图自编码器预测细胞-细胞因子相互作用的生物信息学模型
2022-09-09何瑞瑞朱华庆
朱 渊, 何瑞瑞, 刘 源, 朱华庆*, 李 栋,2),*
(1)安徽医科大学基础医学院, 合肥 230032;2)河北大学生命科学学院, 河北 保定 071002;3)军事医学研究院生命组学研究所,北京蛋白质组研究中心,蛋白质组学国家重点实验室, 北京 102206)
免疫系统中,不同的免疫功能由高度特化的不同类型的细胞执行,各种细胞在免疫系统中的交互作用类似于一个社会网络,而细胞因子则是连接这一网络的重要分子。细胞因子是由免疫细胞和某些非免疫细胞经刺激而合成、分泌的一类具有广泛生物学活性的小分子多肽蛋白质或糖蛋白质。在免疫系统中,免疫细胞通过交换细胞因子实现细胞间的动态通讯,从而产生各种适应环境的反应。细胞因子包括白细胞介素(interleukin, IL)[1, 2]、干扰素(interferon, IFN)、肿瘤坏死因子(tumor necrosis factor, TNF)[3]、集落刺激因子(colony stimulating factor, CSF)、趋化因子(chemokines)[4]和生长因子(growth factor)[5]等,以自分泌、旁分泌、内分泌3种方式作用于自身细胞或其他细胞,通过结合相应的受体诱导细胞凋亡、调控细胞发育和分化、调控机体免疫应答、介导炎症反应和促进组织修复[6]。众多细胞通过细胞因子在机体内相互促进或相互制约,形成极其复杂的细胞因子调节网络。
目前,已发展了多种实验方法用于发现细胞-细胞因子的信息传递网络[7, 8],但这些方法通常存在周期长、设备要求高和成本高等缺陷,有必要发展生物信息学方法助力细胞因子-细胞相互作用的发现。为此,Kveler等[9]发展了一种文本挖掘的方法,从广泛疾病条件下的所有可用的PubMed摘要中提取340种细胞类型与140种细胞因子的关系,建立了目前规模最大的细胞-细胞因子相互作用数据集immuneXpresso (iX)。进而通过聚类的方式来系统地预测细胞和细胞因子之间的相互作用关系。然而,iX数据集以及[9]文中预测的839对细胞-细胞因子的结果,同整个的免疫细胞-细胞因子相互作用空间(iX数据集提供,包括345种免疫细胞和143种细胞因子)相比仍然是极为有限,有必要发展更为高效的预测模型,进一步推进细胞-细胞因子相互作用的研究。细胞与细胞因子相互作用的预测本质是一种关联关系的研究,变分图自编码器(VGAE)作为一种无监督学习框架,被广泛应用于链接预测任务[10-12],其有望在细胞-细胞因子相互作用的预测中发挥重要作用。
VGAE将变分自编码器(variational auto-encoder, VAE)中的变分思想引入到图,充分利用图在表征和计算领域的优点[13]。图是现实生活中广泛存在的一类数据[14],生物医学领域中的分子网络、化合物分子等都可以用图来表示。利用VGAE中的图卷积神经网络(graph convolutional neural network, GCN)能够很好地融合图的网络拓扑特征和节点属性特征[15, 16]。除此之外,自编码器因其较强的特征学习能力、训练速度快、更少的中间参数等优点而被广泛使用[17]。
因此,本文提出一种基于VGAE预测细胞-细胞因子相互作用的深度学习模型——DeepCKI。此模型利用细胞因子组成的蛋白质-蛋白质相互作用(protein-protein interaction, PPI)网络和蛋白质不同类型的特征构成图,通过GCN学习图的节点以及节点属性之间的内在规律和更加深层次的语义特征。利用已知的细胞-细胞因子相互作用和学习到节点嵌入向量训练深度神经网络分类器,进而预测新的细胞-细胞因子之间的关联。
1 材料与方法
1.1 细胞-细胞因子相互作用的数据集
本文从iX数据库(http://www.immunexpresso.org)下载了文本挖掘获得的人类细胞-细胞因子相互作用数据集。该数据集包括细胞作用于细胞因子以及细胞因子作用于细胞两大类(Table 1)。其中,细胞因子作用于细胞可进一步细分为:细胞因子正调控细胞(cytokine-cell+)、细胞因子负调控细胞(cytokine-cell-)、细胞因子调控细胞(cytokine-cell)。汇总所有数据集并删除重复项,得到3 345对细胞-细胞因子相互作用。
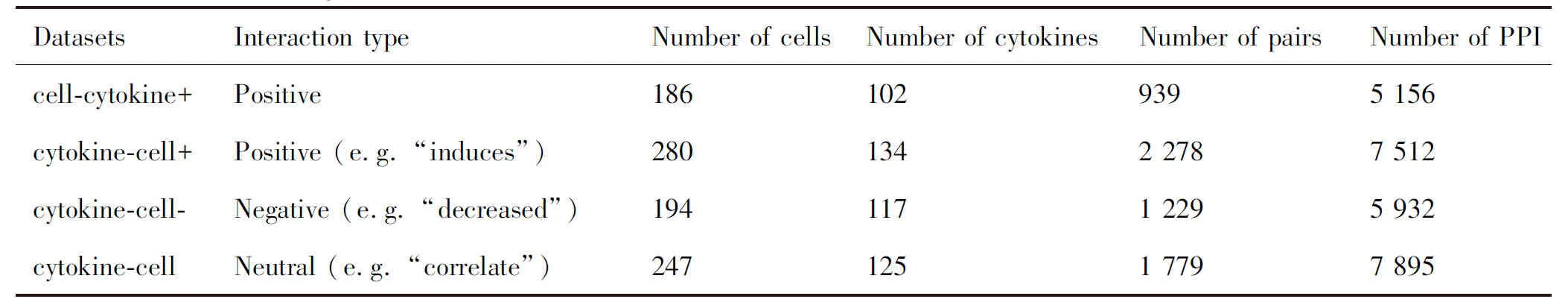
Table 1 Details of cell-cytokine interaction datasets
1.2 图的概念与定义
本文用图来描述蛋白质相互作用网络。图(graph)是由节点(node)和连接这些节点的边(edge)组成的数据结构,图定义为G=(V, E)。其中,V表示节点的集合,E表示节点之间相连边的集合。通用的图表示是一个五元组:G(V, E, A, X, D)。其中,AN×N代表图的邻接矩阵,XN×F代表节点的特征矩阵,DN×N代表度矩阵,N和F分别代表节点的数量和节点特征向量的维度。
1.3 用于预测细胞-细胞因子相互作用的生物学证据
1.3.1 蛋白质-蛋白质相互作用网络 本文从STRING(v11.0)[18]数据库下载PPI(9606.protein.links.v11.0.txt.gz)数据。细胞-细胞因子相互作用数据集划分为不同的类型。根据每种类型数据集中的细胞因子,筛选2个蛋白质都是细胞因子且“combined score”大于300的高可靠的PPI,然后构建邻接矩阵AN×N表示蛋白质之间的连接关系,而如果使用STRING数据库中经过实验验证的PPI,最多只能整合55对PPI,这么小的规模无法构建有效的预测体系,所以,采用了全部证据来源的相互作用。蛋白质vi和vj之间有相互作用,那么对应的邻接矩阵的元素Aij=1,否则Aij=0。邻接矩阵对角元素通常设为0。
1.3.2 蛋白质序列 从Swiss-Prot[19]数据库中下载所有人类蛋白质及其序列数据。采用联合三联体(conjoint triad,CT)[20]编码将蛋白质序列转换为固定维度的嵌入向量,而包含模糊氨基酸(B、O、J、U、X、Z)的蛋白质被删去。常见的20种氨基酸根据偶极子和侧链体积的差异被分成7类(Table 2),所有同一类的氨基酸都被认为是相同的。CT编码嵌入向量的维度为7 × 7 × 7 = 343。任意3个连续的氨基酸作为1个三联体单位,嵌入向量第i维值为三联体在蛋白质序列中出现的频率。

Table 2 Classification of amino acids according to their dipoles and volumes of the side chains
CT定义为:
V=[n0,n1,…,nq-1]
其中,ni是每种三联体在蛋白质序列中出现的频率,q等于343。
1.3.3 蛋白质的亚细胞定位、结构域和功能注释 从Swiss-Prot数据库中下载蛋白质的亚细胞定位(subcellular localization)、结构域(domain)和GO(gene ontology)注释信息,采用词袋模型(bag-of-words)[21]编码上述数据,以蛋白质的亚细胞定位为例,假设本文获得n个不同的亚细胞定位,则亚细胞定位被编码为一个长度为n的二进制向量,向量中的每个元素表示该蛋白质是否被标注了亚细胞定位。对于无任何亚细胞定位注释的蛋白质,它被表示为一个全为0的长度为n的向量。采用词袋模型编码蛋白质结构域和GO条目时,为了避免维度灾难和降低复杂性,只使用在数据集中出现5次以上的结构域条目(term),以及10次以上的GO注释。
1.4 DeepCKI模型架构
DeepCKI整体架构如Fig.1所示。模型主要分为3个模块:输入特征转换模块、VGAE模块和DNN分类器模块。3个模块具体介绍如下:
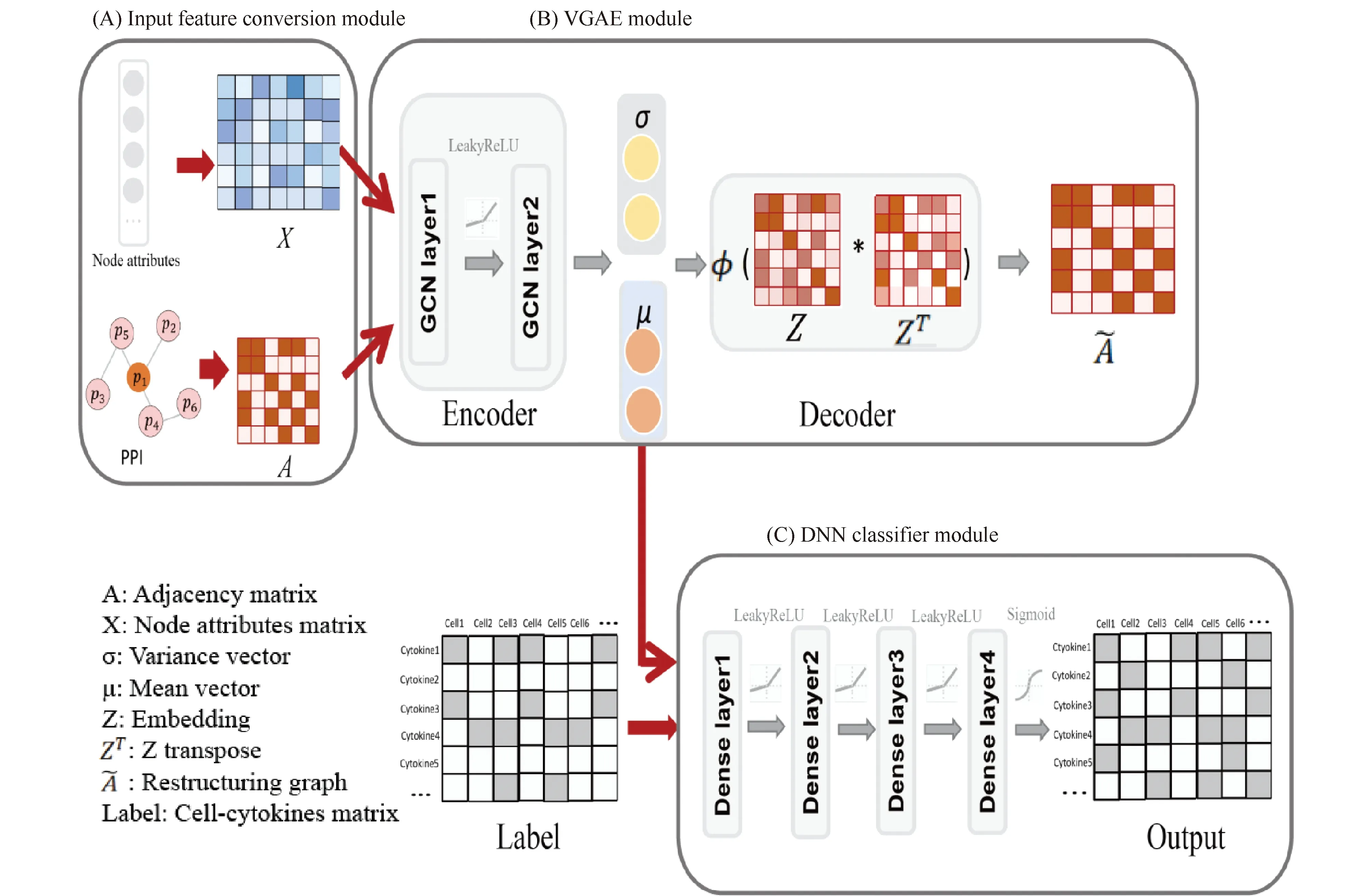
Fig.1 The framework of DeepCKI model (A) Input feature conversion module. This module takes input as the human PPI network downloaded from the STRING database and protein amino acid sequences from the Swiss-Prot database. The PPI network is converted to the format of the adjacency matrix A, and a node attributes matrix X encoded by different protein features. (B) VGAE module. VGAE is an encoder-decoder approach. The encoder is two-layer graph convolutional network and the decoder is a dot product decoder. We obtained the mean embedding vector μ of cytokines for downstream classification tasks. (C) DNN module. The mean vector μ and cell-cytokines label matrix as input for the DNN classification, and the DNN classifier outputs the probabilities of the cell-cytokines interactions
1.4.1 输入特征转换模块 如Fig.1A所示,此模块以 STRING 数据库中下载的人类PPI网络和来自Swiss-Prot数据库的蛋白质氨基酸序列为输入,PPI网络被转换为邻接矩阵A的格式。CT方法编码蛋白质氨基酸序列,词袋模型编码亚细胞定位、结构域和GO 注释,转换后的不同维度的二进制向量作为特征矩阵X。
1.4.2 VGAE模块 如Fig.1B所示,VGAE是一种无监督特征提取方法,它基于网络结构和节点特征生成细胞因子的潜在表示。其一般包含编码器和解码器2个部分,基本思路是:编码器采用2层GCN结构,学习节点隐含向量表示的均值(μ)和方差(σ)的分布,然后从正态分布中采样得到节点的向量表示zi,解码器利用学习到的潜变量Z通过内积的方式重构邻接矩阵A。编码器与解码器的详细构造如下:
1.4.2.1 编码器
VGAE的编码器由GCN组成。它以邻接矩阵A和特征矩阵X作为输入,生成潜在变量Z作为输出。首先,2层GCN定义为:
(1)

Z=μ+σ⊙
(2)
q(zi|X,A) =N(zi|μi,diag(σi2))
(3)
1.4.2.2 解码器
VGAE解码器将使用学习到隐变量zi的内积来重构邻接矩阵:
(4)

1.4.2.3 损失函数

L=Eq(Z|X,A)[logp(A|Z)-
KL(q(Z|X,A)||p(Z)]
(5)
1.4.3 DNN分类器模块 正如Fig.1(C)所示,DNN分类模块由3部分组成:输入层(input layers)、隐藏层(hidden layers)与输出层(output layer)。DNN分类器的输入是每个细胞因子经过GCN编码后提取的均值向量μ和已知的细胞-细胞因子相互作用关系。隐藏层与输出层包含功能神经元,神经元之间的“连接权”,以及每个功能神经元的阈值会在学习过程中根据训练数据进行调整。DNN分类器是一个多标签分类模型,输出为m×n形状的概率矩阵,每一行表示某个细胞因子与n个细胞发生相互作用的概率,每一列表示m个细胞因子与某类细胞发生相互作用的概率。基于概率矩阵与细胞-细胞因子构成的标签矩阵,计算出各个阈值下预测结果的真阳性率和假阳性率,进而绘制n条ROC(Receiver Operating Characteristic)曲线,每条曲线对应一类细胞与m个细胞因子的二分类结果。DeepCKI代码的编写参考了Graph2GO[22]文章。
1.5 预测模型的性能评估
1.5.1 k-倍交叉验证 为了验证模型的效果,本文将所有已知细胞-细胞因子相互作用的数据集数据分为k份,k-1份作为训练集,1份作为验证集,依次轮换训练集和验证集k次。交叉验证可降低由于训练集和验证集单次划分而导致的偶然性,提高泛化能力。
1.5.2 ROC曲线 ROC的全称是“受试者工作特征”曲线[23],该曲线能够反映在不同阈值下敏感度与特异度之间的关系。一个理想的预测模型应该同时具有较高的真阳性率(true positive rate, TPR)和较低的假阳性率(False Positive Rate, FPR)。ROC曲线图中,每个点以对应的FPR值为横坐标,以TPR值为纵坐标。ROC曲线下面积(area under the curve of ROC, AUC)衡量模型优劣的一种评价指标。本文使用机器学习的Scikit-learn[24]包中的roc_curve和auc函数绘制ROC曲线,并计算AUC值。
1.5.3 精确率、召回率和F1-score 真阳性(true positive, TP)指的是将阳性样本正确预测为阳性样本;假阳性 (false positive, FP)指的是将阴性样本错误预测为阳性样本;真阴性(true negative, TN)指的是将阴性样本正确预测为阴性样本;假阴性(false negative, FN)指的是将阳性样本错误预测为阴性样本。本文同时使用精确率(precision)、召回率(recall)和F1-socre评估模型的预测性能。具体计算公式如下所示:
(6)
(7)
(8)
2 结果与讨论
2.1 细胞-细胞因子预测模型DeepCKI的建立
本文通过整合细胞因子的PPI网络和蛋白质序列特征,建立了细胞-细胞因子预测模型——DeepCKI,该模型包含3个模块,其中输入特征转换模块实现蛋白质相互作用网络和蛋白质属性特征到编码向量的转换;VGAE模块使用Adam优化器进行迭代地更新神经网络权重,学习率lr等于0.001,迭代次数epochs等于60,2层GCN隐藏层神经元个数分别是800和400;DNN模块的隐藏层为3层,每层神经元的个数分别为1 024、512、256,在训练过程中对每层的输入数据加1个批标准化处理(batch normalization),隐藏层的激活函数为LeakyRelu,添加Dropout防止模型过拟合,Adam优化器实现对模型的优化,二进制交叉熵损失(binary crossentropy)作为损失函数,输出层使用sigmod激活函数完成最终的多标签分类任务。本文利用已知细胞-细胞因子相互作用数据集训练DeepCKI模型,训练的目标是不断调节模型的超参数,提高模型预测的准确率,降低损失函数的值。DeepCKI基于Tensorflow深度学习框架,代码和数据集都已上传到https://github.com/zhuyuan804/DeepCKI/tree/master。
2.2 特征选择对DeepCKI模型预测效果的影响
为了验证不同类型的特征对模型预测性能的影响,进而寻找最合适的特征或特征组合。本文使用蛋白质序列、亚细胞定位、结构域、GO 功能4种蛋白质属性特征来训练模型,从中选择与细胞因子信息传递功能最相关的特征。其中,不同特征的嵌入向量以首尾横向拼接的方式实现特征的组合。5倍交叉验证(5-fold cross-validation)评估模型预测效果,ROC曲线下面积评估蛋白质不同属性特征对DeepCKI模型预测性能的影响。
由Fig.2A可知,在只使用单类型特征的情况下,蛋白质序列作为特征时模型的ROC曲线下面积等于0.8701,优于亚细胞定位、结构域、GO功能。在Fig.2(B)中,将序列特征分别与亚细胞定位和结构域进行组合,模型预测性能略低于只使用序列信息。虽然GO和序列的组合对模型的预测效果略有提升,但可以忽略不计。总之,预测结果说明,蛋白质序列特征比其他类型特征包含更多的信息量,多种特征的组合并不能显著提升模型的预测性能,而仅使用序列特征就能达到,较好的预测效果。因此,本文仅使用蛋白质序列特征对细胞-细胞因子相互作用进行预测。
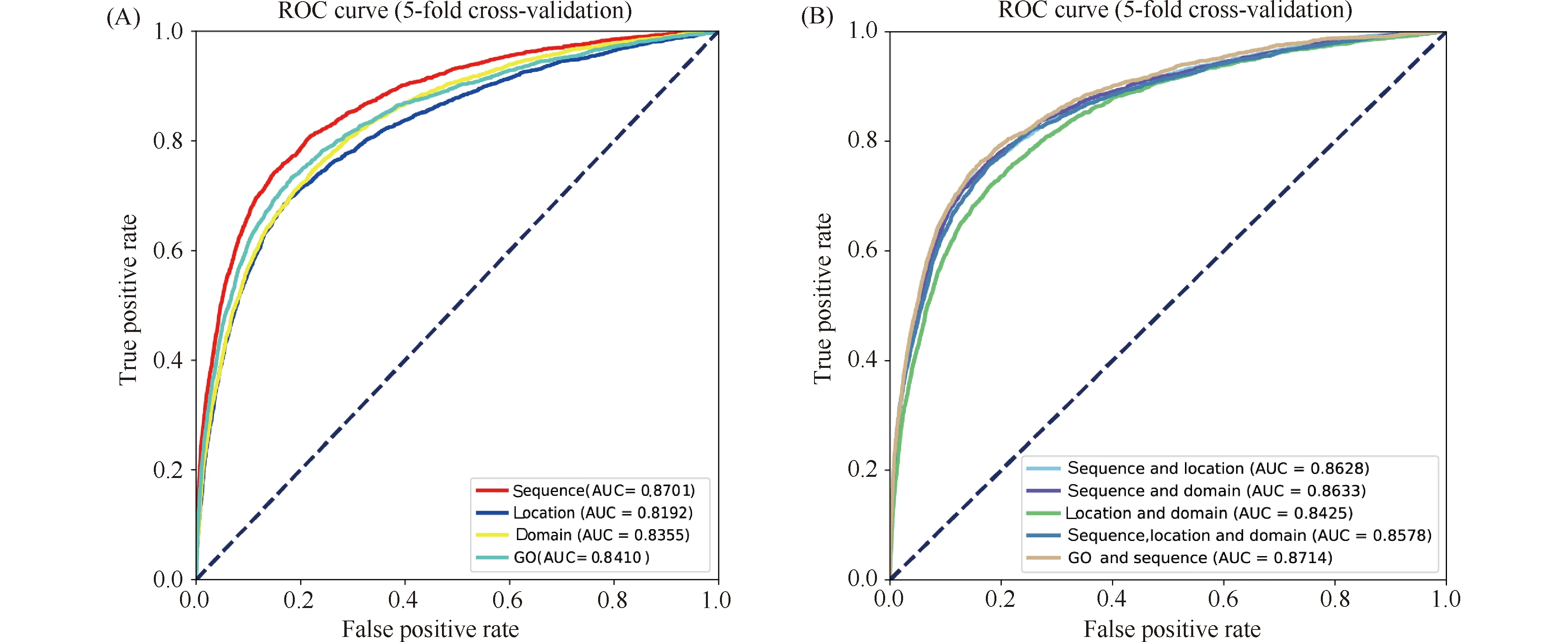
Fig.2 Prediction model performance comparison against using different protein attributes (A) Individual protein attributes sequence, subcellular location, protein domains and GO. (B) Combined protein attributes. sequence and location, sequence and domain, location and domain, sequence, location and domain, GO and sequence
2.3 DeepCKI模型与两类经典深度学习模型预测效果的比较
VAE与DNN是两类经典的深度学习算法,可以用于链接预测或分类任务。VAE无法读入图,DNN仅是对细胞-细胞因子相互作用数据进行简单的分类训练,并不能捕捉细胞因子之间的内在相似性,而DeepCKI可以用于图,因此,本文建立的DeepCKI模型在预测细胞-细胞因子相互作用时,将会比上述2种经典深度学习模型更有优势。为了验证这一假设,本文利用相同的去除重复的3 345对细胞-细胞因子相互作用数据集对3个模型的预测性能进行比较,并利用5倍交叉验证评估模型预测性能。
从Fig.3的结果可知,基于变分图自编码器的DeepCKI模型取得了最优的结果(AUC=0.8701),远高于VAE模型(AUC=0.7637)和DNN模型(AUC=0.6960)。DeepCKI模型预测性能得到显著提高的原因是因为VAGE相对于VAE而言增加了对图特征的提取,编码器的GCN结构从输入的网络拓扑结构和蛋白质序列中捕获细胞因子包含的深层次信息。结果可知,将VGAE学习到细胞因子的隐含表示均值向量μ输入到DNN分类器,比简单使用DNN进行分类更为有效。
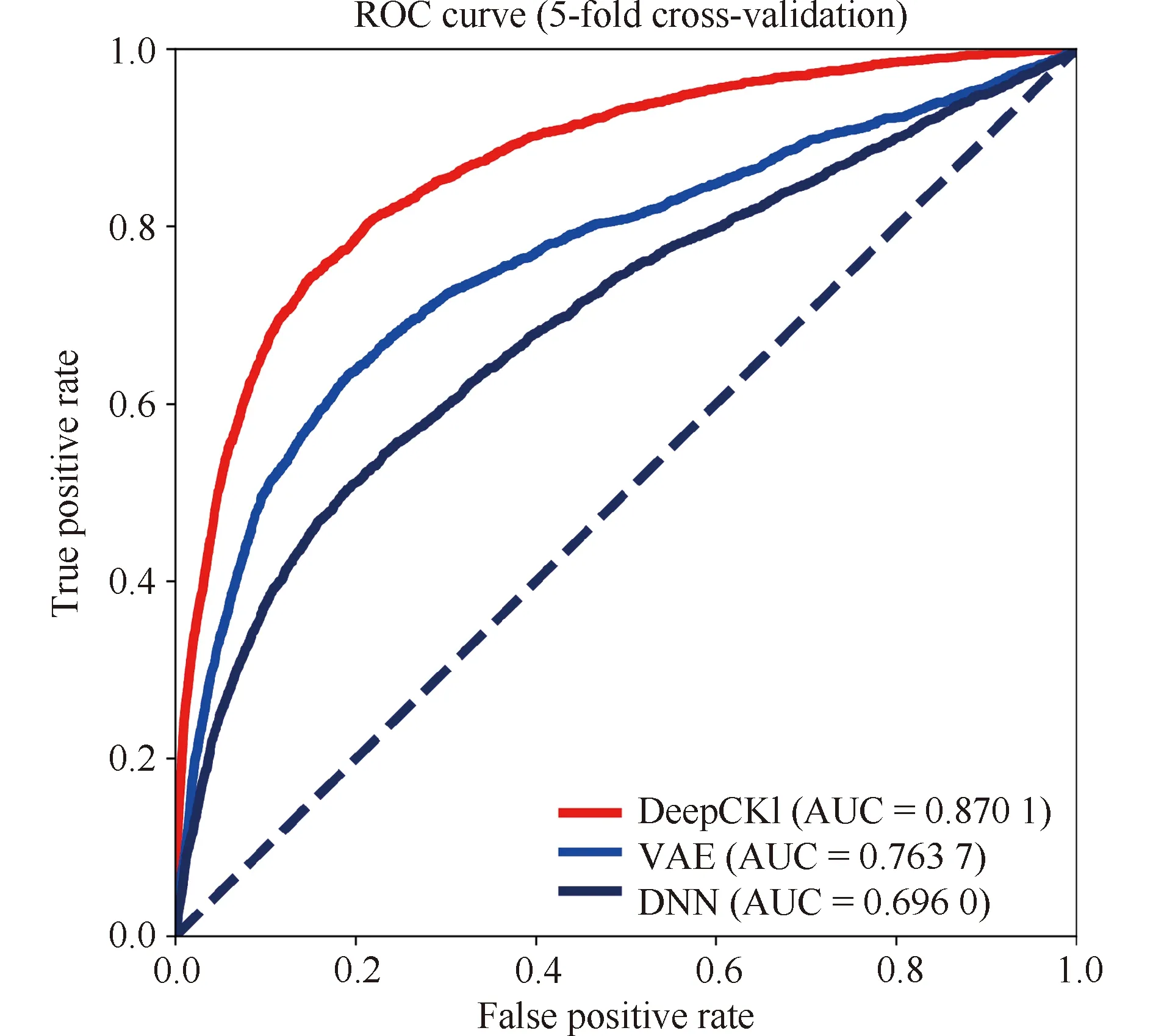
Fig.3 Comparison of DeepCKI, VAE and DNN Each model was evaluated using 5-fold cross-validation. DeepCKI has the highest area under ROC curve (AUC=0.8701) among the three models
2.4 DeepCKI模型在不同类型细胞-细胞因子数据集上的表现
细胞-细胞因子之间具有多种类型的关联关系(详情可查看Table 1),其中包括:细胞正调控细胞因子(例如,巨噬细胞和T细胞产生IL-10促进淋巴细胞性脉络丛脑膜炎病毒(LCMV)克隆感染的慢性[25])、细胞因子正调控细胞(例如IL-2在体外能有效诱导T细胞扩增[26])、细胞因子负调控细胞(例如IL-21和IL-12抑制宫颈癌患者调节性T细胞的分化[27])、细胞因子调控细胞(例如IL-7, IL-4, IL-6,IL-10作用于B细胞[28])。为了考察DeepCKI模型预测不同类型细胞-细胞因子预测的能力,本文使用Table 1中不同类型的细胞-细胞因子相互作用数据对DeepCKI模型进行训练,采用AUC、精确率、召回率和F1-score共4种评估指标评估模型预测性能。由Fig.4可知,虽然在拥有最多PPI数据的cytokin-cell得不到最高的召回率和F1-score,但他的精确率和F1-score值最高。模型在4种类型数据集上的AUC值均在0.8 以上,证明了模型具有较好的预测能力。此外,本文发现在,4类相互作用中,已知PPI越多,该类AUC值越高,表明数据集越完整,算法可以学习到更多的信息,预测更加准确。本文使用cell-cytokine+数据集将该模型与上述2种深度学习模型(VAE和DNN),以及17种来源于Pykeen[29]包中基于知识图谱用来预测节点之间关系的嵌入方法。结果正如Fig.5所示,DeepDKI具有最优的预测性能,进一步证明了模型的高效预测性能。
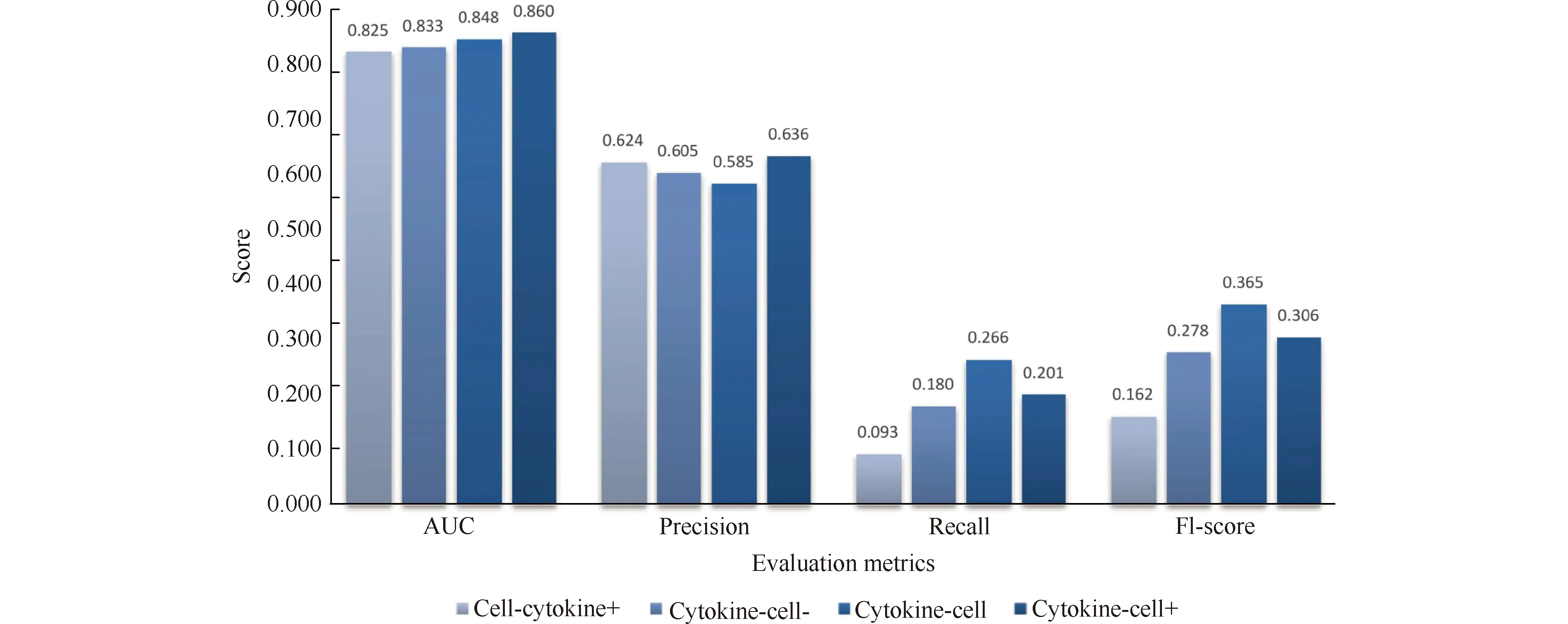
Fig.4 AUC, precision, recall, and F1-Score values of DeepCKI model against four different datasets during 5-fold cross validation The AUC values of the model on the four types of data sets are all above 0.8, which proves that the model has good predictive ability
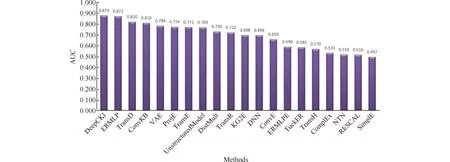
Fig.5 The performance of the DeepCKI and 19 other methods on the cell-cytokine interaction datasets These methods include two deep learning models (VAE and DNN). Seventeen embedding methods from the pykeen package are used to predict relationships between nodes. DeepDKI has the best predictive performance
2.5 DeepCKI模型预测细胞-细胞因子网络
为了进一步验证DeepCKI模型的预测能力,本文使用3 345对细胞-细胞因子相互作用数据集对DeepCKI模型进行训练,在全蛋白质组范围内进行细胞-细胞因子的关联预测。删除与已知数据集重叠的部分,本文预测获得12 410对细胞-细胞因子相互作用(结果未显示)。利用Cytoscape[30]开源软件对预测分值排名前100的细胞-细胞因子相互作用进行可视化展示,结果正如Fig.5展示,涉及21种细胞与41种细胞因子,其中有36对在最新发表的文献中被证实,表明DeepCKI具有发现新的细胞-细胞因子相互作用的关系的能力。
结果正如Fig.6所示,巨噬细胞(macrophage)和T细胞具有最多的细胞因子数量。文献证明,巨噬细胞存在于大部分组织中,是一类具有吞噬作用的天然免疫细胞,参与细胞碎片和病原体的识别、吞噬和降解,在炎症和宿主防御中以及疾病进展中发挥至关重要的作用[31]。当巨噬细胞受到刺激时,通过释放TNF、IL-1、IL-6、IL-20、趋化因子(C-X-C motif)配体9 (chemokine (C-X-C motif) ligand 9, CXCL9)等细胞因子发挥重要调节作用。其中,TNF是引起感染性休克的主要细胞因子之一,在下丘脑中TNF刺激促肾上腺皮质释放激素的释放,抑制食欲,诱导发烧[32]。IL-1是一个多效应的细胞因子,在炎症期间IL-1可刺激肝产生急性期蛋白(acute phase proteins),并作用于中枢神经系统,诱导发热和前列腺素分泌[33]。IL-6具有促炎和抗炎的双向调节功能,影响从免疫到组织修复和新陈代谢的调节过程[32]。IL-20可以通过调节脂肪生成和巨噬细胞失调参与肥胖[32, 34],促进白细胞和上皮细胞之间的细胞通讯[35]。巨噬细胞来源的CXCL9为免疫检查点阻断后的抗肿瘤免疫应答所必需[36]。T细胞是免疫系统中核心组分及效应细胞,具有多种细胞亚型,包括CD8+T细胞,CD4+ T细胞以及T调节细胞等。CD8+T细胞可将细胞毒素释放到受感染的细胞,导致细胞死亡。肿瘤微环境中的CD8+T细胞可生成IL-2、IL-12和IFNγ,靶向杀死肿瘤细胞。CD4+ T细胞分泌的CCL2可召集免疫调节细胞在感染部位集聚。调节性T细胞在活化时可分泌免疫调节因子,例如IL-10、转化生长因子β和IL-35。不同细胞因子也参与调节T细胞的功能,CXCL1通过调节T细胞功能来帮助宿主防御微生物败血症[37],CCL8诱导活化的T细胞趋化[37, 38],MIF(macrophage migration inhibitory factor)抑制T细胞活化[39]。细胞因子IL23A可以同最多类型的细胞关联。一方面,细胞因子IL-23A可以由多种免疫细胞分泌,例如巨噬细胞[40],CD4阳性T细胞(CD4-positive T cell)[41]和B细胞[42]。另一方面,IL-23A可以调控多种细胞的功能,例如可使极化T细胞向不同的效应功能表型分化,参与活化T细胞增殖的正向调节,抑制调节性T细胞活性,导致T细胞依赖性结肠炎[43]。Fig.5网络的分析结果与文献报道一致,其一定程度上证明了DeepCKI模型预测结果的可靠性。
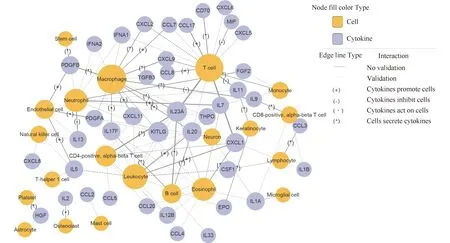
Fig.6 Insights into the top 100 predicted cell-cytokine interaction network Orange and purple nodes, respectively, stand for cell and cytokine. Edges represent cell and cytokine interactions. Solid lines indicate that the predicted cell-cytokine interactions have been validated in the literature, and dotted lines indicate that they have not been validated. " (+)” denotes cytokines that promote cells, e.g. induces"; "(-)" indicates that cytokines inhibit cells, e.g. "decreased"; "(·)" means cytokines act on cells, a neutral regulation, e.g. "correlate”; (*) indicates that cells secrete cytokines
了解免疫细胞间网络的通讯对理解疾病中的免疫反应至关重要。本文构建的预测细胞-细胞因子相互作用的深度学习模型加速推进对细胞间通讯的系统研究。该模型采用图结构设计,以细胞因子组成的邻接矩阵A和蛋白质序列组成的特征矩阵X作为输入,通过整合网络节点和节点特征生成潜在表示,以DNN分类器输出预测的细胞-细胞因子相互作用。通过对不同蛋白质特征进行筛选,发现仅使用序列作为初始特征时模型的预测性能最优,说明蛋白质序列特征比其他类型特征包含更多的信息量。此外,具有GCN结构的DeepCKI模型性能,优于无GCN结构的变分自编码器和不经过编码直接使用特征信息进行分类的深度神经网络,说明GCN能从图中学习到节点和边的内在规律,以及节点属性蕴含更加深层次的特征信息。此外,该模型在不同类型数据集的训练中均表现出了鲁棒性和有效性,并且新预测的细胞和细胞因子,通过查阅文献证明其与多种疾病的发生发展密切相关。
综上结果可以推测,DeepCKI模型具有发现新的细胞-细胞因子相互作用的能力,有助于为大规模的细胞-细胞因子的实验研究提供一定的理论指导。但模型尚不能实现对细胞-细胞因子激活/抑制关系及方向性的预测。未来,可在此方面进一步拓展模型的性能,同时对模型的可解释性进行深入研究。
猜你喜欢
杂志排行

中国生物化学与分子生物学报的其它文章
- 半翻转生物化学教学模式对学生学业成绩和自我认知的影响
- 人多能干细胞来源的肾定向分化平台的构建
- 泛素样蛋白UBQLN2通过抑制结肠癌Wnt信号通路发挥抑癌作用
- 两种表达系统来源新冠病毒RBD蛋白免疫小鼠后的IgG抗体水平对比研究
- Potent Anti-bacteria Activity of Tamsulosin Hydrochloride and Ningmitai Capsule in Combination by Regulating the Gene Expression Related to β-Lactamase, Hemolysis, and Virulence
- 利拉鲁肽抑制高糖诱导的小鼠胰岛细胞损伤