污水中常见毒品的分析方法优化及验证
2022-09-07杭太俊王优美
王 叶,徐 磊,徐 鹏,杭太俊,宋 敏*,王优美,徐 慧
(1中国药科大学药学院,南京 210009;2国家禁毒委员会办公室中国药科大学禁毒关键技术联合实验室,南京 210009;3公安部禁毒情报技术中心,北京 100741)
毒品严重影响人类健康,破坏家庭和睦,危及社会公共安全。毒品的种类、来源、吸毒及致死人数不断发生变化,据统计,全球约有2.7 亿人使用过毒品,我国现有吸毒人员约180 万,滥用合成毒品人员103 万[1-2]。全球毒品滥用问题错综复杂,造成了严重社会危机。
污水流行病学方法[3-4]根据毒品吸食后经过代谢,以原型或代谢物的形式通过尿液、粪便等排入城市污水处理系统,通过检测污水处理厂污水中毒品及其代谢物的浓度,结合污水流量、毒品代谢率、社区人口数等数据评估社区范围内毒品的滥用情况[5-10]。与传统的毒情检测手段相比,基于污水分析法的毒情评估具有客观、定量、普遍、实时、快速简便等优点。
污水中毒品含量低,由于稀释效应,浓度一般为ppt 级,且干扰成分多,对样品前处理和分析检测技术有较高的要求[11-13]。根据目标物的理化性质选择合适的前处理方法,并采用高灵敏度分析仪器,是实现准确定性定量分析的基本前提。
现常采用固相萃取技术(SPE)对污水样品进行浓缩,然后利用LC-MS/MS 对污水样品进行定量测定,常采用同位素内标进行定量[14-15]。国内外文献对于有机氮碱性物质的研究洗脱液多选择含有一定比例氨水的甲醇或乙腈溶液洗脱,然后洗脱液在40~50 ℃的条件下氮气吹干或减压离心浓缩挥干,最后复溶,进LC-MS/MS分析[16-19]。本实验室前期根据国内外已有的污水分析法,建立了基于SPE-UPLC-MS/MS 分析方法对污水中多种滥用药物及其代谢物的定量分析方法[20]。然而在实际应用过程中有机氮碱性物质稳定性较差,内标响应偏低,低浓度点的准确性和重复性较差,故方法仍需进一步优化。
本研究对已有的SPE-UPLC-MS/MS 分析方法在实际应用过程中存在的问题进行分析,优化了样品前处理的关键条件,在样品浓缩过程中加酸酸化,使目标物呈盐酸盐的形式,保障了针对这些有机氮碱性痕量成分分析的抗交叉污染性,提高了有机氮碱性物质污水分析方法的稳定性、专属性和准确性。建立了城市污水中包含苯丙胺和吗啡在内的12种常见毒品及其代谢物的痕量分析方法,并成功地应用于城市毒情评估。
1 材 料
1.1 试药与试剂
12 种毒品、毒品代谢物的标准品以及各自相应的氘代内标储备液均购于美国Cerilliant 公司,包括:吗啡(MOR)、6-单乙酰吗啡(6-MAM)、苯丙胺(AM)、甲基苯丙胺(MAM)、亚甲氧基苯丙胺(MDA)、二亚甲基双氧苯丙胺(MDMA)、甲卡西酮(MC)、可卡因(COC)、苯甲酰爱康宁(BZE)、可待因(COD)、氯胺酮(KET)、去甲氯胺酮(NK)、MOR-D3、6-MAM-D3、AM-D5、MAM-D5、MDA-D5、MDMA-D5、MC-D3、COC-D3、BZE-D3、COD-D6、KET-D4、NK-D4。
甲醇、乙腈(CR 级,德国Merck 试剂);甲酸、盐酸、氨水(AR 级,南京化学试剂有限公司);去离子水(市售娃哈哈纯净水)。
1.2 仪 器
超高效液相色谱-串联质谱联用仪(色谱仪:美国Thermo Scientific Vanquish UPLC,质谱仪:美国Thermo Scientific TSQ Quantis);24位固相萃取装置(美国Supelco 公司);Oasis Prime MCX 固相萃取柱(美国Waters公司);减压离心浓缩仪(美国Labconco公司);百万分之一天平(美国Mettler Toledo公司)。
2 方 法
2.1 检测条件
2.1.1 色谱条件 采用Zorbax Eclipse Plus C18色谱柱(50 mm × 2.1 mm,1.8 μm),流动相A 为0.1%甲酸水溶液,流动相B 为乙腈,线性梯度洗脱(A∶B):0 min(95∶5)→6 min(75∶25)→6.2 min(0∶100)→8 min(0∶100)→8.1 min(95∶5)→11 min(95∶5)。流速为0.3 mL/min,柱温为40 ℃,进样盘温度为4 ℃,进样量为10 μL。
2.1.2 质谱条件 离子源为电喷雾离子源(ESI源),检测方式为ESI+,毛细管电压为3.5 kV,鞘气流速为35 Arb,辅助气流速为5 Arb,尾吹气流速为0 Arb,离子源传输管温度为350 ℃,雾化气温度为50 ℃,扫描方式为MRM,根据以往研究[20]和质谱扫描结果确定各目标物的MRM监测通道。
2.2 溶液配制
2.2.1 混合对照品溶液 取6-MAM、AM、MAM、MOR、MDMA、MDA、MC、KET 和COC 标准品约2 mg,精密称定,分别置于10 mL 量瓶中,其中,6-MAM、MOR、MC 和COC 用乙腈溶解并稀释至刻度,其余目标物用甲醇溶解并稀释至刻度,摇匀,配制成质量浓度约为200 μg/mL的上述各目标物的储备液。精密吸取BZE、NK 和COD 1 mg/mL的标准品溶液1 mL,分别置于10 mL 量瓶中,用甲醇稀释至刻度,摇匀,配制成质量浓度为100 μg/mL的上述各目标物的储备液。将上述储备液逐级稀释,配制成上述目标物质量浓度为2.5 μg/mL(AM为6.25 μg/mL)的混合对照品溶液。
2.2.2 混合内标溶液 精密吸取各氘代内标100 μg/mL的标准品溶液1 mL,分别置于10 mL 量瓶中,其中MC-D3、6-MAM-D3和COC-D3用乙腈稀释至刻度,其余内标用甲醇稀释至刻度,摇匀,分别配制成质量浓度为10 μg/mL的上述各目标物的储备液。将上述储备液逐级稀释,配制成质量浓度为25 ng/mL的氘代同位素内标混合溶液。
2.3 样品前处理
2.3.1 样品采集与过滤 采集污水厂进水口格栅过滤后未处理的污水样品;样品为24 h 混合样(每1小时采1次,每次100 mL,最后混合摇匀),用干净聚丙烯瓶盛装,加入浓盐酸调节pH 小于2.0,-20 ℃保存。分析前将污水样品融化,并用玻璃纤维滤膜过滤。
2.3.2 固相萃取 取过滤后水样50 mL,加入混合内标溶液100 μL,充分振摇混合均匀,以约 4 mL/min的速度上样Oasis Prime MCX 固相萃取柱,上样完成后,先用甲醇4 mL 淋洗萃取柱,再用5%的氨水/乙腈溶液4 mL 洗脱(流速约为1 mL/min)并收集洗脱液,在50 ℃条件下减压离心浓缩至约0.2 mL,加盐酸-乙腈(5∶95)20 μL,混匀后减压离心浓缩挥干,残渣物用甲醇-水-甲酸(5∶95∶0.1)溶液100 μL 复溶,离心取上清液进UPLC-MS/MS分析。
2.4 方法学考察
2.4.1 标准曲线和范围 取混合对照品溶液适量,用甲醇逐级稀释,分别配制成含AM 1.25,6.25,12.5,62.5,125,312.5,625 ng/mL;其他目标物含0.5,2.5,5,25,50,125,250 ng/mL的系列标曲混合溶液。
取不同浓度的系列标曲混合溶液100 μL,混合内标溶液100 μL,分别用50 mL 纯水稀释,配制成含AM 分别为2.5,12.5,25,125,250,625,1 250 ng/L;其他目标物为1,5,10,25,50,100,250,500 ng/L的模拟样品。按“2.3.2”项进行试验,记录色谱图,以各目标物的峰面积比值Y(= As/Ar)对目标物浓度X(ng/L)进行权重(1/X2)回归分析。结果表明,AM 在2.5 ~ 1 250 ng/L 范围内,其余目标物在1 ~ 500 ng/L 范围内,相关系数(r)均大于0.99,质谱响应呈良好线性关系。
2.4.2 定量下限 按“2.4.1”项下,配制LLOQ的模拟样品(AM 为2.5 ng/L,其他目标物为1 ng/L),平行6份,按“2.3.2”项下方法进行试验,根据当日标准曲线计算每一样本测得浓度。结果表明(表1),重复测得的精密度和准确度均符合LLOQ的要求。故本方法中AM的定量下限可达到2.5 ng/L,其余目标物的定量下限可达到1 ng/L。
2.4.3 残留效应 按“2.4.1”项下,配制标曲最高浓度的模拟样品(AM 为1 250 ng/L,其他目标物为500 ng/L),平行6份,按“2.3”项下方法进行试验,记录各目标物及其内标的峰面积。结果表明(表1),高浓度样品之后在空白样品中的残留不超过定量下限的20%,并且不超过内标的5%。故残留效应对水样中各目标物测定的影响可以忽略不计。
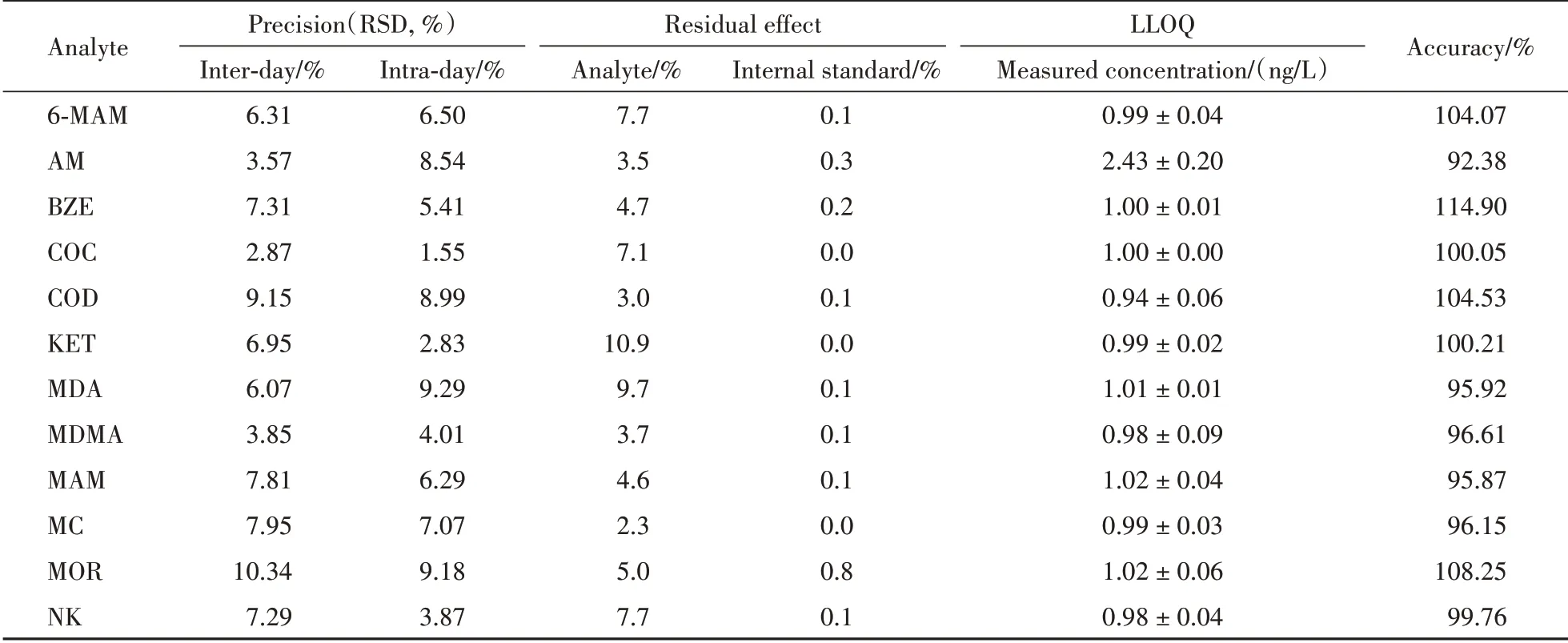
Table 1 Precision,residual effect,lower limit of quantitation(LLOQ)and accuracy of the method
2.4.4 回收率和基质效应 取污水基质50 mL,按“2.4.1”项下,配制成低(L)、中(M)、高(H)3 个浓度的质控样品(AM 为5、125、1 000 ng/L,其余目标物为2、50、400 ng/L)各6份,按“2.3.2”项下方法平行操作,作为回收率样品组(基质效应对照组)。另取污水基质50 mL,加入甲醇100 μL,进行固相萃取操作,向洗脱液中加入L、M、H 3 个浓度的混合溶液100 μL 和混合内标溶液100 μL,每一浓度各6份,按“2.3.2”项下方法起操作,作为回收率对照组。取洗脱液4 mL,加入L、M、H 3 个浓度的混合溶液100 μL 和混合内标溶液100 μL,每个浓度平行3份,按“2.3.2”项下方法起操作,作为基质效应对照组。记录各目标物及其对应内标峰面积,扣除污水基质中各待测物峰面积后,计算各目标物的回收率和基质效应。结果(表2)表明,各目标物的回收率和基质效应均符合要求。
2.4.5 准确度和精密度 取污水基质50 mL,按“2.4.1”项下,配制成LLOQ、L、M、H 4 个质量浓度的质控样品(AM 为2.5、5、125、1 000 ng/L,其余目标物为1、2、50、400 ng/L)各6份,按“2.3.2”项下方法平行操作。根据当日标准曲线计算每一样本实际浓度,一个分析批内平行测定多次(n=6),计算批内变异。不同分析批测定多次(n= 3),计算批间变异。结果(表1)表明,各目标物的准确度和精密度均符合要求。
2.5 检测方法在实际污水样品中的应用
利用优化后的SPE-UPLC-MS/MS 分析方法对东部某市14 家污水处理厂的污水样本进行分析,并根据检测结果反算各毒品的平均消耗量。结果表明,所建立的样品前处理方法及检测条件能准确检测12种常见毒品及其代谢物在污水厂进水中的浓度,满足实验要求。
3 讨 论
3.1 SPE条件优化
已有SPE 方法中[21-23],洗脱液在50 ℃条件下挥干浓缩过程中未加酸酸化,在应用中发现AM、MAM 和MC的内标响应异常偏低,导致目标物低浓度点的准确性和重复性较差。分析不加酸时回收率实验中相同浓度的内标溶液的响应值,发现差异较大,AM、MAM 和MC 3 个目标物内标响应的RSD分别为52.75%、61.60%和101.40%。

Table 2 Recovery,matric effect of the method
氘是自然界存在的氢同位素,氘代化合物是将化合物分子中某个或某些C-H 键中的氢原子替换成氘原子。研究表明,氘代化合物的沸点和未氘代化合物的沸点存在微小差异[24],如一个标准大气压下丙酮的沸点为56 ℃,氘代丙酮的沸点为55.5 ℃。因此,推测AM、MAM 和MC 与氘代内标的沸点存在的差异导致了内标响应值变化。由于AM、MAM、MC的pKa均大于7,为碱性物质,当洗脱液呈碱性时,这3种物质及其氘代内标均为游离分子状态,在减压离心浓缩过程中易挥发,而目标物和内标的沸点不同,导致测定结果准确性和重复性较差,且低浓度点特别明显。
因此,在样品减压离心浓缩至相对较少体积时,加入盐酸-乙腈(5∶95)进行酸化,使AM、MAM和MC 及其内标转化为更加稳定、不易挥发的盐酸盐,减少目标物和氘代内标的挥发,使内标响应稳定,AM、MAM、MC 3 个目标物的氘代内标响应值RSD 分别为27.80%、20.13%和18.61%。加酸酸化保障了针对这些有机氮碱性痕量成分分析的抗交叉污染性,提高了方法的稳定性、专属性和准确性(表3)。

Table 3 Comparison of measured concentration with or without acid
3.2 待测物的选择依据
在进行污水流行病学分析时,需要选取一种或几种代谢产物作为该毒品代谢目标分析物(DTR)进行测定,从而反推出该毒品的消耗量。DTR 多选取最主要、量最大、在污水中稳定性良好且易于监测的毒品代谢物[7]。根据以往研究对国内 毒 品 滥 用 情 况的 研 究[2,20],本研 究 选择 监 测MOR、6-MAM、AM、MAM、MDA、MDMA、MC、COC、BZE、COD、KET、NK 12 种毒品及其代谢物。其中MOR 和6-MAM 作为海洛因的DTR,MAM 作为冰毒的DTR,BZE 和COC 作为COC的DTR,MDMA 和MDA 作为摇头丸的DTR,MC 作为MC的DTR,AM作为AM的DTR,KET 和NK 作为KET 当然DTR,COD作为COD的DTR。
由于污水中AM的来源较多,AM、冰毒及其他AM 类物质均可代谢产生AM[25-26],同时根据实际样本浓度确定AM的线性范围高于其他目标物。
3.3 东部某市14家污水处理厂检测结果分析
对东部某市14家污水处理厂的污水样本分析结果显示,MOR、MAM 和KET的 浓 度 较高,6-MAM、BZE、COC 均未检出。6-MAM 为海洛因的代谢物,海洛因在体内水解成6-MAM,而6-MAM 很快就代谢成MOR[27],故海洛因仅有极少量(约0.5%)代谢为6-MAM 排出体外[6],故在污水中很难检出。BZE 是COC 在体内主要代谢物[28],两者均未检出说明COC在该市并不存在滥用。
在实际污水样本测定过程中发现,个别污水样品中个别目标物的内标通道无响应,推测是污水样品中存在氧化剂,内标加入后被氧化。故在污水样品抽滤后加入适量的还原剂(巯基乙醇),结果表明随着还原剂浓度的增加,内标响应恢复,表明个别污水样品中存在氧化剂影响分析结果的准确性。
4 结 论
本研究对已有的污水样品前处理方法进行优化,在挥干过程中加酸调节pH,可以减少AM、MAM 和MC的挥发,通过方法学验证,证明优化后的SPE-UPLC-MS/MS 检测方法高效可靠。对东部某市14 家污水处理厂的污水样本测定结果表明,优化后的前处理方法和UPLC-MS/MS 检测方法能够满足分析要求。
