心-面-皮肤综合征合并耳聋患儿基因变异1例报道
2022-09-01孙淑妮闫露露楼碧莹薛江阳郝雯颖李海波
孙淑妮 闫露露 楼碧莹 薛江阳 郝雯颖 李海波
患儿女,9个月,因“生长发育迟缓”于2020年9月13日就诊于宁波市妇女儿童医院儿科门诊。患儿孕34周出生,出生体重2 560 g,出生身长48 cm,新生儿听力筛查及脑干听力诱发电位未通过,串联质谱未见异常,运动、语言发育落后,9个月还坐不稳。患儿母亲有不良孕产史:胚胎停育1次,未行遗传学检查;孕24+2周时超声提示羊水偏多(图1a)、胎儿四肢长骨异常(图1b-c),行羊水穿刺后染色体核型和基因芯片检测结果未见明显异常;家族中无类似病史,父母体健,父方有耳聋家族史:患儿爷爷耳聋,姑姑目前出现听力下降,否认近亲婚配及其他家族遗传病史。体格检查(图2):神志清,特殊面容,包括额头突出、低耳位、长脸、长人中、头发稀疏卷曲,眼距宽、眼裂小、双眼斜视,朝天鼻、手外翻,发绀,漏斗胸,肌张力低下,房间隔缺损,重度营养不良,严重发育迟缓。头颅CT检查提示双侧脑实质肿胀,脑部MRI检查提示双侧大脑半球脑白质内片状稍长T1稍长 T2信号。

图1 产前超声及四维彩超检查结果(a:羊水偏多;b-c:四肢长骨偏短)

图2 患儿临床表现(a:颈后组织增厚,头发稀疏卷曲;b:眉毛稀疏,额头突出、低耳位、长人中,朝天鼻;c:漏斗胸,手外翻;d:小下颌,手外翻,四肢纤细,肌张力低下)
经患儿家长同意后进行基因检测,采用外显子捕获技术,使用Novaseq测序平台(美国Illumina公司)对基因的外显子及侧翼的内含子区域进行测序分析。全外显子组测序及Sanger测序验证结果:在该患儿中检出MAP2K1基因c.383G>T(p.Gly128Val)杂合变异(图3a),父母未检出该变异,提示为新发变异(PS2+PS4_P+PM1+PM2+PP3)。同时在该患儿中检出GJB2基因c.109G>A(p.V37I)和 c.187G>T(p.V63L)复合杂合变异(图3b-c),分别遗传自父亲和母亲。根据美国遗传学与基因组学会(ACMG)基因变异分类指南,c.109G>A为致病性变异(PS4+PM3+PP1_S),c.187G>T为临床意义不明确变异(PM2+PP3)。结合临床和基因检测等资料,该例患儿诊断为心-面-皮肤综合征(cardio-facio-cutane‐ous syndrome,CFC)。本研究经宁波市妇女儿童医院医学伦理委员会批准(EC2020-048),患儿双亲均签署知情同意书。
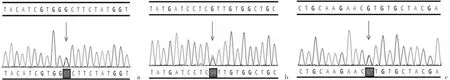
图3 患儿MAP2K1和GJB2基因Sanger测序结果[a:患儿携带c.383G>T错义变异(■标记处);b:患儿GJB2基因存在c.109G>A变异;c:患儿GJB2基因存在c.187G>A变异)]
讨论CFC是一组先天性多发性异常疾病,其主要临床特征为特殊面容、心脏缺陷、智力低下、生长发育迟缓等[1-2]。CFC具有遗传异质性,多个致病基因突变均可导致CFC,其中CFC 3型(cardio-faciocutaneous syndrome 3,CFC3,MIM:612279)是由MAP2K1基因缺陷所致。
MAP2K1基因位于人类15号染色体长臂上(15q22.1-q22.33),基因全长104 kb,包含11个外显子[3]。MAP2K1基因编码MEK1蛋白激酶,该蛋白激酶是有丝分裂原激活蛋白激酶信号转导途径中关键的组成成分,主要参与细胞增殖、分化、基因转录调控和发育过程[4]。Reynolds等[5]在1986年最早报道了该疾病。人类基因组突变数据库(HGMD)已收录19例MAP2K1基因变异,且变异集中在第2、3、6外显子区[6]。
据文献报道,MAP2K1基因变异所致的CFC3患儿出生前后会有相应的临床表现。在产前,患儿超声下可呈现多种表现,如颈部透明层增加、水囊瘤、短股骨、胎儿水肿以及羊水指数>25 cm[7-8]。出生后,CFC3可累及全身多个系统,主要的异常面容包括前额宽大、上睑下垂、鼻基底部宽、低位耳等,同时可有头面部发育畸形伴小下颌畸形、高颚弓等;眼部异常,如斜视、眼球震颤等;对于皮肤病变,可见多发的色素痣、黑棘皮病等表现[9];循环系统方面异常,包括肺动脉瓣狭窄、房室间隔缺损等;神经系统方面,CFC可导致患儿智力发育障碍,部分患儿会出现癫痫症;消化系统方面,CFC可引起喂养困难,包括胃食管反流、吞咽障碍等[10];颅脑结构及功能的异常,包括脑白质发育不良、脑间隙增宽、脑电图出现异常;另外还可累及运动系统、生殖泌尿系统等[11]。此外,国内外文献报道还提及罕见的临床表型,如喉气管软化、声门下重度狭窄及巨大眼睑裂缝孔[12-13]。本例患儿除了表现出CFC3典型的临床特征(如特殊面容、肌张力低下、喂养困难、发育迟缓等)之外,还表现出漏斗胸。Aoki等[14]报道CFC患儿由于氧化磷酸化功能障碍,肌肉内辅酶Q10缺乏容易并发漏斗胸、脊柱侧弯、关节挛缩、扁平足等,本例患儿除漏斗胸外无上述其他表现。
本例患儿产前超声显示羊水过多、颈部半透明增加以及长骨发育异常。Myers等[15]报道产前颈部淋巴结结构异常、羊水异常及长骨发育不良是CFC3患儿的相关表现,并且出生后会经历早期吞咽障碍和喂养困难,该患儿产前超声检查结果与本例患儿相似。然而,张欢欢等[4]报道了1例与本例患儿MAP2K1基因相同变异的患儿,临床表现为反复癫痫发作,心电图、脑电图提示异常,但未提示心脏畸形、胸廓畸形及无产前B超羊水过多、颈部半透明层增加和长骨发育异常,与本例患儿不同。
本例患儿除发现有MAP2K1基因变异外,还伴有GJB2基因变异,这两种基因同时变异致CFC3发病的情况尚无报道。在我国,GJB2基因变异是遗传性耳聋的主要致病原因之一,本例患儿GJB2基因存在 c.109G>A(p.V37I)和 c.187G>T(p.V63L)复合杂合变异,分别遗传自父亲和母亲,本家系父方有耳聋遗传史,患儿新生儿听力筛查及脑干听力诱发电位未通过。有研究表明,p.V37I纯合子、p.V37I与其他GJB2基因致病变异的复合杂合变异可导致多种表型的听力损失,与迟发性耳聋密切相关[16-18]。另外,文献报道CFC患儿常见外耳畸形(耳位低下、外耳廓前后翻转等),该畸形表现是导致患儿听力损失的主要原因[11,19]。本例患儿存在迟发性耳聋相关的致病基因变异,但目前耳聋尚不能确定由CFC所致。
本研究通过全外显子组测序技术,发现患儿 MAP2K1基因 c.383G>T(p.Gly128Val)变异合并GJB2基因c.109G>A和c.187G>T复合杂合变异,国内外尚未报道,这丰富了CFC3的疾病胎儿表型谱和致病变异谱,对后续遗传咨询和疾病治疗具有重要意义,且说明了产前超声检查可发现胎儿结构异常,指导临床医生及时对新生儿进行干预,做好产前超声诊断可降低新生儿出生缺陷发生率,降低围生儿和婴幼儿死亡率,这对保障儿童健康有着重要的意义。
