严重程度不同的SHANK2 相关神经发育障碍一家系4 例基因型-表型关系研究
2022-09-01田茂强束晓梅雷文婷余小华彭龙英
田茂强 束晓梅 李 娟 雷文婷 陈 静 余小华 彭龙英
遵义医科大学附属医院小儿内科(贵州遵义 563003)
神经发育障碍(neurodevelopmental disorders,NDDs)是一组表现为运动、智力、语言、社交及兴趣异常的神经系统发育障碍谱系病,包括自闭症谱系障碍(autism spectrum disorder,ASD)[1]。此组疾病临床异质性强,虽多数患者与遗传相关,但其基因型-表型关系复杂[2]。目前报道NDDs相关基因变异多数因涉及突触转录调控、影响突触传递功能而致病[3-5]。SHANK2基因(OMIM* 603290)编码SH3和多个锚蛋白重复结构蛋白2(SHANK 2),是一种兴奋性突触后支架蛋白,参与神经兴奋的传导过程,研究发现SHANK2与NDDs 相关[1,5-6]。SHANK2相关NDDs临床表型差异较大,其基因型-表型关系及遗传模式复杂,发病率及病例数不详,亟待进一步研究明确。本研究对父母分别携带SHANK2单位点变异、而下一代携带双等位变异的家系患者的临床表型及遗传学进行分析,以提高临床医师对SHANK 2相关谱系病基因型-表型关系的认识。
1 病例资料
先证者(II.3)为2岁患儿,女,因发育落后就诊。表现为全面发育落后,1岁6个月可抬头,2岁仍不会独坐,不会讲话,可认识亲人。围生期无异常,父母非近亲结婚。家系中共4 例患者表现为不同程度的智力、运动及社会功能障碍。系谱图及相应的临床表型见图1。父亲(I.1)表现为轻度智力障碍,伴社交功能障碍,但生活自理能力正常,能完成日常农活;母亲(I.2)表现为中度智力障碍,伴严重语言及社交障碍,但可完成日常家务活动;大姐(II.1)智力运动发育正常;二姐(II.2)4.5岁就诊,表现为自幼严重的智力运动发育障碍,随访到5岁,仅可认识亲人,不会走路,不会说话。体格检查(先证者2岁时):身长 83 cm,体重 11 kg,无特殊外观,反应迟钝,追声、物好,无语言表达,心肺腹部无异常,四肢肌张力高,病理征阴性。辅助检查:血常规、血生化、血氨、乳酸、丙酮酸正常,叶酸、维生素B12均正常;血、尿串联质谱筛查无异常。II.2及II.3Gessel发育量表评估均提示重度全面发育落后。2 例患儿头颅磁共振成像(MRI)无异常。因经济原因父母未进行发育量表及头颅MRI 检查。因家系中有多例类似患者,考虑遗传性病因可能性大。经医院医学伦理委员会批准、患儿监护人签署知情同意书后,采集家系先证者及父母外周血送至北京贝瑞和康医学检验实验室有限公司进行全外显子测序(WES)。WES检测到SHANK 2基因2 种变异。①SHANK 2剪切位点变异(NM_012309.4,c.2309-2A>C),经Sanger验证显示母亲(I.2)、二姐(II.2)及先证者(II.3)均携带该变异,父亲及大姐该位点为野生型(图2A),该变异在数据库中均未被收录;根据ACMG 评级[7],该位点变异为致病变异(PVS1+PM2+PP1)。②检测到父亲及先证者SHANK2基因11-16号外显子杂合缺失(图3A),经qPCR验证显示父亲(I.1)、二姐(II.2)及先证者(II.3)均携带该缺失,见图3 B 及表1。该家系诊断为SHANK2相关NDDs,诊治时间轴见图4。2例患儿不规则康复治疗中,智力及运动功能缓慢进步中。
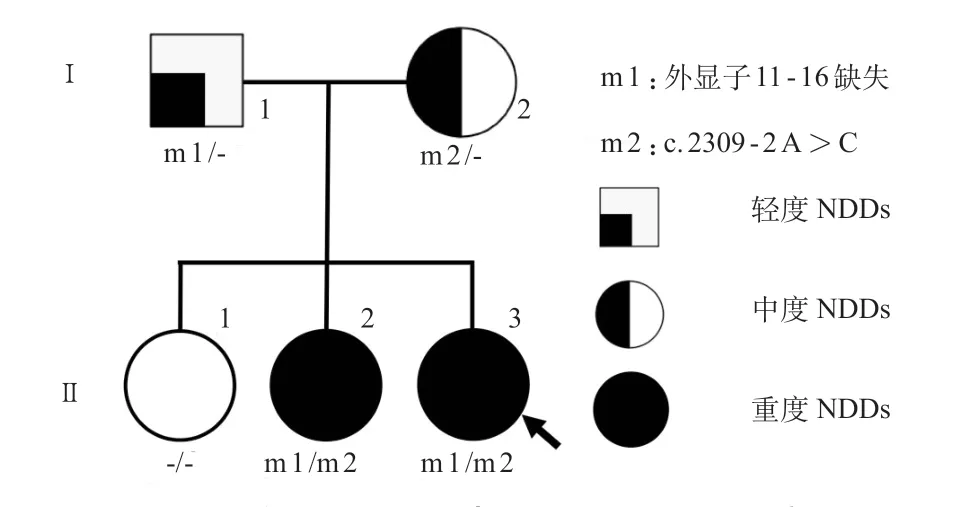
图1 家系系谱图及患者基因型-表型示意图

表1 3例患者qPCR检测结果

图2 家系成员一代测序峰图、SHANK2 结构及其基因型-表型关系示意图

图3 SHANK2 缺失及qPCR 验证图
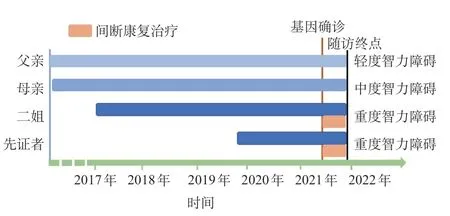
图4 家系患者诊治时间轴
2 讨论
在个体生命发育过程中,神经元突触的密度、大小、形状等在相应基因的调控下处于动态变化中[8-9]。编码这些突触蛋白的基因如SHANK1~3变异均可导致突触功能异常而引起NDDs。SHANK 2包含SH 3、PDZ 结合域及SAM 结构域(图2)[9]。SHANK2(NM_012309.4)含25个外显子、1 849个氨基酸,是SHANK突触蛋白家族(SHANK1~3)成员之一,在兴奋性后突触密度中起到分子支架的作用[10]。SHANK2在脑中高表达,出生前(胎儿期)及成人期高表达,基因敲除小鼠显示产后致死、树突形态异常及兴奋性突触后电位异常(https://gdap.org.cn/unigene.html?entrez=22941),提示SHANK2对维持中枢神经系统的正常发育及功能起至关重要的作用。
SHANK 2基因型-表型关系复杂。在鼠的研究中发现,编码SHANK家族蛋白(SHANK1~3)基因的变异导致与人类NDDs 相似表型,包括重复刻板运动、社会行为改变及焦虑症状[11]。临床研究发现,SHANK2与神经发育障碍相关,包括ASD易感17型(OMIM# 613436),遗传模式为常染色体显性遗传伴不完全外显[5]。SHANK 2复杂的基因型-表型关系可能与单基因的变异类型、修饰基因或多基因遗传相关。本研究家系中,未携带SHANK 2变异的姐姐表型正常,携带缺失变异的父亲表现为轻度社交障碍,携带内含变异的母亲表现为中度智力障碍及社交障碍,而携带双等位基因变异的2 个女儿均表现为严重的运动、认知发育障碍,提示SHANK 2不同变异导致SHANK 2 蛋白剂量的变化可能是该家系患者表型差异的主要原因,也提示SHANK 2谱系病临床异质性可能与变异导致SHANK 2 剂量变化相关。
Zhang等[12]在关于先天性脊柱侧弯2家系研究发现父亲及2个儿子均携带TBX6缺失变异,但仅其中一个儿子表现为脊柱侧弯,即携带变异的个体呈现不完全外显现象。进一步研究发现,发病者还携带来自母亲TBX6的多态性位点(T-C-A)。该研究认为TBX 6缺失变异和多态性位点共同导致先天性脊柱侧弯,呈非典型隐性遗传模式[12]。该现象在RBM8A相关血小板减少症的研究也曾被报道[13]。SHANK2是ASD的易感基因,大量研究发现某些携带变异者也表现为不完全外显的现象[5],提示可能个体对于该基因剂量依赖较低。当变异导致SHANK2轻微下降时则不出现表型或出现较轻微表型,而严重变异或双等位基因变异则导致SHANK2蛋白显著下降而出现较重表型。推测本研究患者中差异性表型出现的原因可能为:①父亲携带杂合外显子缺失变异导致SHANK2截短,机体存在截短的SHANK2及50%功能正常的SHANK2,患者为轻表型(图2);②母亲携带内含子变异(c.2309-2A>C),该变异导致可变剪切,可能导致外显跳跃(缺失)、外显子延长及或外显子部分缺失等,与缺失相比,母亲剪切位点变异可能导致体内存在大量无功能的变异型蛋白,由于显性负效应机制,大量变异型蛋白可能与野生型蛋白SHANK2竞争性的结合靶蛋白,影响残存正常蛋白功能,从而导致比父亲较重的表型(图2);③2个女儿均携带双等位基因变异,则体内仅存在变异型SHANK 2,患儿则表现为重型NDDs(图2)。本研究家系可能为进一步关于SHANK2致病机制研究提供参考,但仍需在今后通过直接的功能实验或积累更多类似病例进行证实。
ASD 是一类以广泛的社交、言语交流障碍、刻板行为及兴趣狭隘为核心症状的综合征[14-15]。通过患病的一致率研究发现同卵双生子为30%~99%,异卵双生子为0~65%,说明ASD病因与遗传因素高度相关[2]。近来发表在Cell杂志的研究已经发现了三类变异与ASD 相关,即在多基因变异的基础上,罕见变异及新生变异是ASD 发生的主要因素[14]。Caumes 等[16]2020 年报道2 例ASD 患者,并总结了已报道的9 例SHANK 2基因变异的患儿,11 例患者均为De novo变异,其中10 例患者伴典型ASD 症状,提示De novo变异多见于对于SHANK2剂量影响较大的变异,常导致ASD,而遗传性变异可能对SHANK2蛋白剂量影响相对较小而导致不同程度智力障碍。本研究中SHANK 2单等位基因和/或双等位基因变异出现的表型异质性现象可能为ASD或智力障碍的研究提供依据,对于表型差异较大的家系患者,除考虑表观遗传或多基因变异影响外,还应该考虑双等位基因特殊变异(拷贝数变异,多态性等)导致不典型隐性遗传可能。
总之,SHANK 2相关NDDs 严重程度可能与SHANK 2 量依赖相关,此种量依赖现象也为理解SHANK 2相关NDDs 基因型-表型关系提供帮助。对于临床表现差异大、不完全外显的家族患者,需进一步分析患者中可能存在该基因的其他类型变异。
