超高效液相色谱-串联质谱法测定畜肉中万古霉素残留量及测量不确定度评估
2022-08-30李慧晨张颖颖郭文萍宋永青樊之聪
李慧晨,张颖颖,郭文萍,宋永青,樊之聪
(中国肉类食品综合研究中心,肉类加工技术北京市重点实验室,北京 100068)
万古霉素(分子式CHClNO)属于糖肽类抗生素,作为抗生素的“最后一道防线”,其独特的抗菌机制被用于治疗对其他抗生素无效的严重感染。万古霉素的药渣可作为补充蛋白饲料资源的一部分,并且可作为保健促生长饲料添加剂,用以提高饲料报酬率,因此被广泛用于食用动物的生产养殖中,尽管农业农村部第250号公告中已将万古霉素列为禁用兽药,但仍有养殖户在饲养过程中恶意添加,造成畜肉药物残留的问题。消费者长期食用会产生严重的抗生素耐药性,且万古霉素有严重的耳毒性、肾毒性及其他副作用,给人类的健康造成巨大影响。
目前,万古霉素的测定样品基质主要集中在药物、血液、饲料、乳制品等,采用的方法多是液相色谱法、酶联免疫吸附法、化学发光免疫法、近红外光谱法、微生物法和液相色谱-串联质谱法。万古霉素经过饲喂及体内吸收后,残留在畜肉中的含量较低,超高效液相色谱-串联质谱(ultrahigh performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)具有分辨率高、定性定量准确、检出限低、灵敏度高等优点,能够对畜肉中万古霉素进行痕量检测。近年来对畜肉中万古霉素残留量测定的研究不断增多,回收率和检出限也能满足日常检测需求,但普遍未对方法中基质效应进行评价,也未评估检测过程的不确定度。
食品检验工作对结果的准确度要求日益提高,但检验过程会受到诸多因素的影响,导致测量结果和真值之间存在一定的误差。由于真值往往不易得到,为了更客观、科学地评价测量结果,需要开展对食品检测过程中不确定度的评定。本研究利用UPLC-MS/MS,通过优化样品前处理条件及仪器采集条件,建立一种畜肉中万古霉素的检测方法,并依据CNAS-CL01-G003—2021《测量不确定度的要求》分析方法测量结果的不确定度,以便对用检测结果进行样品符合性判定时的风险开展评估,确保实验质量。
1 材料与方法
1.1 材料与试剂
生鲜猪肉(15 份)、牛肉(5 份)和羊肉(5 份)购买于农贸市场。
万古霉素盐酸盐标准品 德国Dr. Ehrenstorfer公司;乙腈(色谱纯)、甲酸(色谱纯) 美国Fisher公司;正己烷(分析纯)、甲醇(分析纯) 国药集团化学试剂(上海)有限公司。
1.2 仪器与设备
TSQ UPLC-MS/MS仪(配备电喷雾离子源、TraceFinder 4.1数据处理系统) 美国Thermo Fisher公司;20PR-520高速冷冻离心机 日本Hitachi公司;HGC-12A氮吹仪 天津市恒奥科技发展有限公司;HENGAO T&D固相萃取装置 美国Aiglent公司;KQ-700DV超声仪 昆山超声仪器有限公司;C、C、PHENYL固相萃取柱 博纳艾杰尔科技有限公司。
1.3 方法
1.3.1 标准溶液的配制
准确称取0.014 75 g万古霉素标准品(精确到0.000 01 g),用体积分数0.1%甲酸水溶液-乙腈定容于100 mL容量瓶中,制得标准储备液质量浓度为147.5 µg/mL,4 ℃冰箱保存。使用时依次用0.1%甲酸水溶液-乙腈(7∶3,/)稀释至所需质量浓度。
1.3.2 样品制备
提取:分别取猪肉、牛肉和羊肉样品,经匀浆后制备成糜状均质样品,准确称取5 g均质样品(精确到0.001 g)于50 mL离心管中,加入5 mL 0.1%甲酸水溶液-乙腈(70∶30,/)溶液,涡旋2 min后,超声10 min,10 000 r/min离心5 min;上清液全部转移至另一50 mL离心管中,再向样品残渣中加入5 mL 0.1%甲酸水溶液-乙腈(70∶30,/)溶液,重复提取1 次,离心后合并提取液;加入5 mL乙腈饱和的正己烷,涡旋1 min,4 000 r/min离心5 min后,弃去上层正己烷。
净化:将提取液全部加入至依次用5 mL甲醇、5 mL水活化的C固相萃取柱中,用5 mL水淋洗小柱,然后用6 mL甲醇洗脱,收集全部洗脱液,50 ℃氮吹至干;准确吸取1.00 mL 0.1%甲酸水溶液-乙腈(70∶30,/)溶解残渣,涡旋混匀后过0.22 µm滤膜,进行UPLC-MS/MS检测及分析。
1.3.3 仪器条件
液相色谱条件:Hypersil GOLD C色谱柱(2.1 mm×100 mm,1.9 μm);流速0.2 mL/min;柱温25 ℃;进样量10 µL;流动相A:体积分数0.1%甲酸水溶液,流动相B:乙腈。二元梯度洗脱程序见表1。

表1 流动相梯度洗脱程序Table 1 Gradient elution program of mobile phase
质谱条件:离子化模式为正模式;喷雾电压3 000 V;离子源雾化温度350 ℃;鞘气压力45 arb;辅气压力15 arb;采集方式:正离子多反应监测(multiple reaction monitoring,MRM)模式,采集参数见表2。
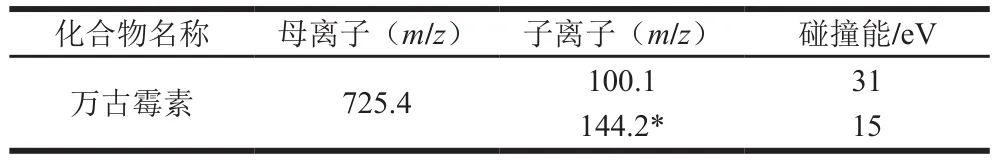
表2 万古霉素的MRM采集参数Table 2 Mass spectrometric parameters for vancomycin
1.3.4 标准曲线绘制
分别准确移取万古霉素标准溶液0.20、0.50、1.00、2.00、5.00 mL,用0.1%甲酸水溶液-乙腈(70∶30,/)定容至50 mL,配制成质量浓度分别为0.59、1.48、2.95、5.90、14.75 ng/mL的标准工作溶液。采用外标法,以待测物质量浓度为横坐标,以待测物定量离子峰面积为纵坐标,绘制标准曲线。
1.3.5 测量不确定度评定
对猪肉中万古霉素含量进行6 次重复测定,根据测定结果,依据CNAS-CL01-G003—2021和JJF1059.1—2012《测量不确定度评定与表示》,对测定过程产生的不确定度进行评估。
1.3.6 基质效应评价
基质效应按式(1)计算。

式中:基质匹配标准曲线以空白基质提取液为溶剂,配制成与标准曲线各点质量浓度相同的标准工作溶液,绘制标准曲线。
1.4 数据处理
色谱图采用Thermo Fisher TraceFinder 4.1进行数据采集和处理,自动计算拟合线性回归方程。
2 结果与分析
2.1 样品前处理条件优化
2.1.1 提取溶液的选择
万古霉素属于大环糖肽类抗生素,化学结构中含有羟基、酰胺等多个亲水性基团,在水溶液中有很好的溶解性,在甲醇中微溶,在中性和酸性溶液中较稳定。实验比较了水、体积分数0.1%甲酸水溶液、体积分数0.3%甲酸水溶液的提取效果。结果显示,酸性水溶液提取率高于水溶液,且0.1%甲酸水溶液与0.3%甲酸水溶液的提取率无明显差别,故选择0.1%甲酸水溶液进行提取。但由于畜肉中有大部分蛋白质是溶于水的,提取液浑浊,杂质较多,基质干扰严重且容易污染质谱,根据畜肉中蛋白质特点,加入适量的有机溶剂可降低蛋白质的溶解性,因此比较在甲醇、乙腈中分别加入0.1%甲酸水溶液的提取率,结果显示,0.1%甲酸水溶液-乙腈的提取率最高,因此选用0.1%甲酸水溶液-乙腈作为混合提取试剂。
进一步比较不同体积比的0.1%甲酸水溶液-乙腈(85∶15、70∶30、55∶45)对样品中万古霉素的提取回收率,结果显示,0.1%甲酸水溶液-乙腈(70∶30,/)的回收率最高。此外,畜肉中所含的脂肪会干扰净化过程并污染色谱柱,因此提取溶液需用乙腈饱和的正己烷去除脂肪后再进一步净化。
2.1.2 净化条件的选择
固相萃取柱的选择:根据万古霉素的化学性质,分别选择C、C、PHENYL 3 种固相萃取柱进行净化,结果显示,C萃取柱的回收率最高,因此选择C作为固相萃取柱。
净化条件的优化:比较水和0.1%甲酸水溶液作为淋洗溶剂的效果,结果显示,二者回收率无明显差别,因此选择水作为淋洗溶剂;进一步比较3、6、10 mL甲醇作为洗脱溶剂的效果,结果显示,用3 mL甲醇洗脱回收率最低,用6、10 mL甲醇洗脱的回收率相差不大,因此选择6 mL甲醇进行洗脱。
2.2 仪器条件的优化
万古霉素的分子式为CHClNO,相对分子质量为1 449.25,对万古霉素标准物质进行全扫描时,发现万古霉素更容易产生带正电的双电荷离子([M+2H]),因此其母离子为/725.4。之后进行质谱条件的优化,通过优化碰撞电压等条件,获得定量离子、定性离子的最大响应强度。正离子扫描模式下,在液相色谱流动相中加入少量的甲酸会增强待测化合物的响应强度,这主要是由于酸提供了更多的H。因此本实验选用0.1%甲酸水溶液与乙腈为流动相,优化梯度洗脱程序,从而保证各个化合物有良好的峰形、分离度及响应强度。在此条件下,10 ng/mL万古霉素标准溶液的定量离子色谱图如图1所示。
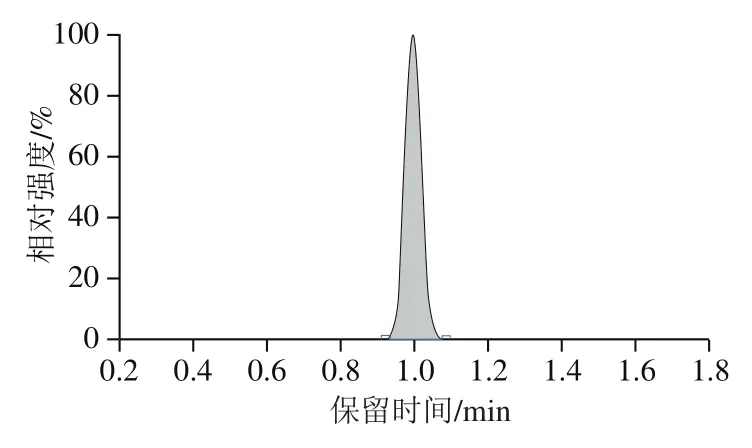
图1 万古霉素标准溶液色谱图Fig. 1 Chromatogram of vancomycin standard solution
2.3 方法学验证
2.3.1 线性范围及检出限
在0.59~14.75 ng/mL质量浓度范围内,线性方程为=94.19+142.50,相关系数为0.999 7,线性关系良好。
在空白猪肉样品中加入万古霉素标准溶液,经过提取和净化后,进行仪器检测,以定量离子信噪比不小于3为检出限,得到检出限为0.1 µg/kg。较周迎春等的研究,该方法检出限更低。
2.3.2 回收率及精密度
在空白猪肉中添加低、中、高3 个浓度水平的标准溶液,每个添加量分别做6 个平行,进行回收率实验。由表3可知,万古霉素的回收率为86.7%~100.3%,相对标准偏差小于3%,其精密度和回收率均符合残留分析方法的要求。在此基础上,对空白牛肉、羊肉进行低浓度水平(加标量为0.38 µg/kg)的添加回收验证实验,回收率分别为86.3%和97.1%,满足日常检测需求。

表3 猪肉样品中万古霉素添加回收率及相对标准偏差Table 3 Recoveries and RSDs of vancomycin in spiked pork samples
2.3.3 基质效应评价
基质效应是影响定量分析准确性的因素之一,产生的主要原因是样品中盐类、胺类、脂肪酸等干扰物质与目标物一同提取出来,从而影响目标物进入质谱分析的离子数量,导致定量结果产生偏差。本研究中万古霉素的基质效应为-24%,结合添加回收实验结果,综合认为基质效应未对实验结果产生显著影响。
2.4 方法的应用
根据本实验建立的前处理方法,对农贸市场的猪肉、牛肉、羊肉共计10 个样品进行万古霉素含量检测,未检出阳性样品。
2.5 不确定度评估
由于既往对万古霉素检测方法的研究中,测量不确定度评估的研究较少,因此,本研究对猪肉中万古霉素测定过程的测量不确定度进行了评估。
2.5.1 建立数学模型
样品中万古霉素含量按式(2)计算。

式中:为试样中万古霉素含量/(µg/kg);为从标准工作曲线中拟合得到的万古霉素溶液质量浓度/(ng/mL);为样品溶液最终体积/mL;为样品质量/g。
2.5.2 不确定度分量的主要来源分析
通过检测过程及数学模型,可以确定不确定度来源主要包括标准溶液配制过程引入的不确定度、样品制备过程引入的不确定度、曲线拟合引入的不确定度、重复性实验(随机)变化引入的不确定度、试剂空白引入的不确定度、提取、净化对样品回收率的影响引入的不确定度和仪器定量测量重复性引入的不确定度。
相对合成标准不确定度按式(3)计算。

2.5.3 标准溶液配制过程引入的不确定度
来自标准储备液配制过程引入的不确定度和标准溶液配制过程引入的不确定度,按式(4)计算。

2.5.3.1 标准储备液配制过程引入的不确定度
来自标准物质纯度引入的不确定度、标准物质称量引入的不确定度和标准储备液定容至100 mL时引入的不确定度。按式(5)计算。
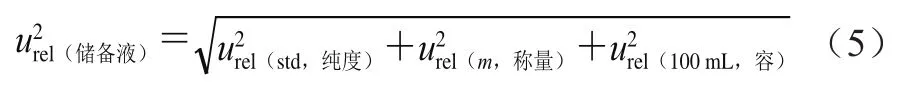
式中:标准品证书上提供的标准物质纯度为(99.5±0.5)%,包含因子为3,则按式(6)计算。
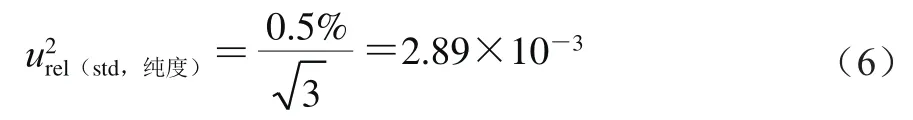
天平检定证书给出的最大允许误差为±0.01 mg,包含因子为3,则按式(7)计算。
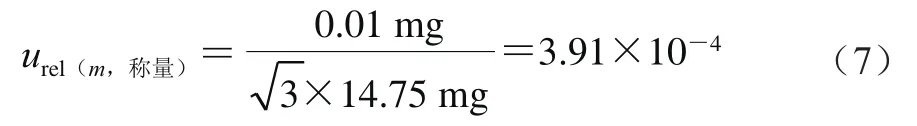
分别来源于容量瓶引入的不确定度和实验温度引入的不确定度。100 mL容量瓶的允许误差为±0.1 mL,包含因子为6;实验温度为22 ℃,水的体积膨胀系数为2.1×10/℃,包含因子为3。因此,按式(8)计算。

根据式(8)计算得到为4.74×10,根据式(5),利用式(6)、(7)、(8)的结果,计算得到为2.95×10。
2.5.3.2 配制标准溶液引入的不确定度
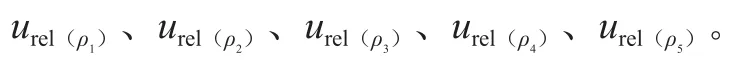
来自0.2 mL分度吸管引入的不确定度和定容引起的不确定度,按式(9)计算。
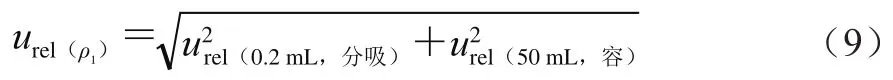
式中:0.2 mL分度吸管的允差为±0.005 mL,50 mL容量瓶的允差为±0.05 mL,包含因子均为6,实验温度引入的不确定度同上,则和分别按式(10)、(11)计算。

根据式(10)、(11)计算得到为1.02×10,为4.74×10。
根据式(9),利用式(10)、(11)的结果,计算得到为1.02×10。
来自0.5 mL分度吸管引入的不确定度和定容引起的不确定度,按式(12)计算。
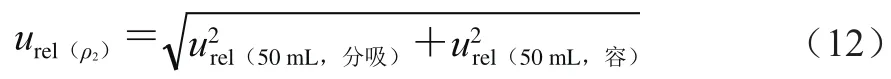
式中:0.5 mL分度吸管的允差为±0.010 mL,包含因子为6,实验温度引入的不确定度同上,按式(13)计算。

根据式(13)计算得到为8.16×10。
根据式(12),利用式(11)、(13)的结果,计算得到为8.17×10。
来自1.0 mL单标吸管引入的不确定度和定容引起的不确定度,按式(14)计算。

式中:1.0 mL单标吸管的允差为±0.007 mL,包含因子为6,实验温度引入的不确定度同上,按式(15)计算。

根据式(15)计算得到为2.87×10。
根据式(14),利用式(11)、(15)的结果,计算得到为2.91×10。
来自2.0 mL单标吸管引入的不确定度和定容引起的不确定度,按式(16)计算。

式中:2.0 mL单标吸管的允差为±0.010 mL,包含因子为6,实验温度引入的不确定度同上,按式(17)计算。

根据式(17)计算得到为2.05×10。
根据式(16),利用式(11)、(17)的结果,计算得到为2.10×10。
来自5.0 mL单标吸管引入的不确定度和定容引起的不确定度,按式(18)计算。

式中:5.0 mL单标吸管的允差为±0.015 mL,包含因子为6,实验温度引入的不确定度同上,按式(19)计算。

根据式(19)计算得到为1.24×10。
根据式(18),利用式(11)、(19)的结果,计算得到为1.33×10。
根据式(4),利用式(5)、(9)、(12)、(14)、(16)、(18)的结果,计算得到标准溶液配制过程引起的相对标准不确定度为1.39×10。
2.5.4 样品制备过程引入的不确定度
来自称量引入的不确定度和用单标吸管移取1.0 mL甲酸-乙腈溶液复溶所引入的不确定度,按式(20)计算。
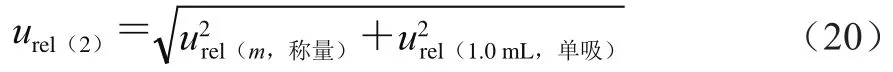
式中:称取5.000 g样品,天平检定证书给出的最大允许误差为±0.001 g,包含因子为3,则按式(21)计算。
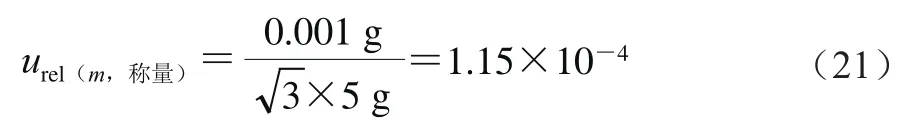
根据式(20),利用式(15)、(21)的结果,计算得到样品制备过程引入的不确定度为2.87×10。
2.5.5 曲线拟合引入的不确定度
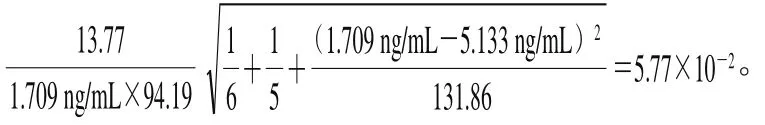
采用5 个质量浓度水平的万古霉素标准溶液(ρ)进行线性拟合,得到线性方程:=94.19+142.50(=0.999 5),曲线拟合的相对合成不确定度按式(22)计算。
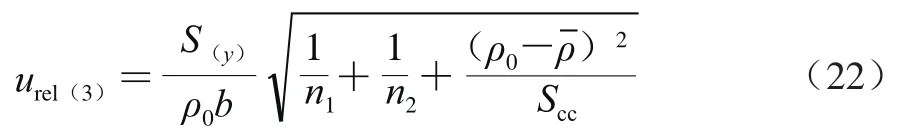
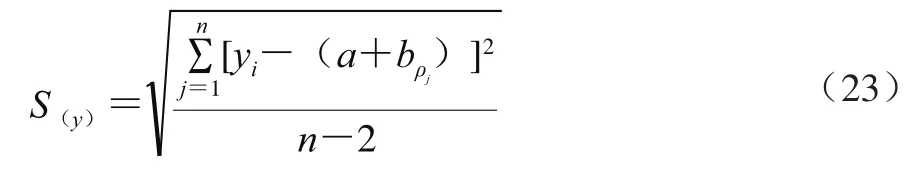
式中:为拟合曲线截距(142.50);y为拟合曲线各个质量浓度的响应值;ρ为拟合曲线各质量浓度。

2.5.6 重复性实验引入的不确定度
在重复性条件下,对猪肉中的万古霉素含量进行了6 次平行测定,含量(x)分别为0.341、0.350、0.367、0.326、0.348、0.319 µg/kg。重复性实验的相对合成不确定度按式(25)计算。


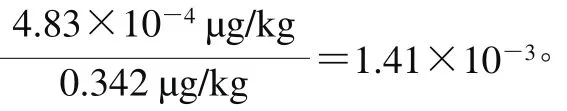
2.5.7 试剂空白引入的不确定度
由于本实验所用试剂均为色谱纯或者分析纯,试剂空白溶液在目标物保留时间附近无干扰,故可忽略不计。
2.5.8 提取、净化对样品回收率的影响引入的不确定度
对含有万古霉素的样品进行6 次添加回收实验,加标回收率分别为88.8%、91.1%、88.8%、86.7%、89.8%和89.6%。加标回收率实验的相对合成不确定度按式(27)计算。

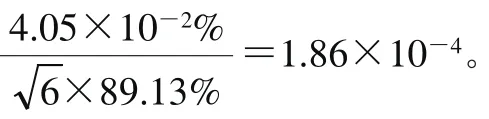
2.5.9 仪器定量测量重复性引入的不确定度
根据校准证书获得仪器定量测量重复性引入的不确定度=6.12×10。
2.5.10 相对合成标准不确定度

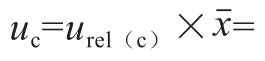
3 结 论
本研究建立了测定畜肉中万古霉素残留的UPLCMS/MS法。样品经0.1%甲酸水溶液-乙腈提取,正己烷除去脂肪,过C固相萃取柱净化,C色谱柱分离,采用MRM检测,外标法定量。检出限为0.1 µg/kg,加标回收率为86.7~100.3%,相对标准偏差小于3%。对该过程的测量不确定度进行评估,在95%的置信水平下,取扩展因子=2,结果可表示为(0.342±0.041) µg/kg。本方法操作简便快捷、灵敏度高、回收率稳定,数据准确,满足国内外相关法规的要求,适合大批量样品的分析。
