超高效液相色谱-串联质谱法同时测定动物源性食品中9 种多肽类抗生素残留
2022-08-27李莲微杨晓聪姚闽娜刘正才李小晶
李莲微,杨晓聪,姚闽娜, ,刘正才,李小晶
(1.福建农林大学食品科学学院,福建福州 350002;2.信阳农林学院食品学院,河南信阳 464000;3.福州海关技术中心,福建福州 350001)
多肽类抗生素是一类具有多肽结构特征的抗生素的总称,通常含有15~45 个氨基酸残基[1],并且分子量大、结构复杂,它们中的大多数通过破坏微生物的细胞膜结构诱导微生物死亡以实现抗菌功能,少数直接穿透细胞膜并与不同的靶点结合发挥抗菌作用[2],因此被广泛用于动物饲养中作为饲料添加剂和兽药使用,以促进动物生长和预防疫病[3]。动物生产过程中抗生素的违规使用和滥用一方面破坏了抗菌药物和细菌耐药性之间的平衡,加快了细菌耐药菌的产生进程[4],这些耐药菌通过环境和食品加工环节在动物源性食品中传播,临床上的“超级细菌”和“超级耐药基因”逐渐蔓延并出现在食品中,很可能导致人类生病后无药可治[5-7]。另一方面也带来了食品安全隐患,长期摄入这些动物产品,食物中残留的多肽类抗生素也会对人体产生一定的毒副作用[8]。此外,该行为还会打破动物体内的微生态平衡,造成动物免疫力减退[9],累积的兽药以及其代谢物被排泄到土壤和水中,也可能对农业生态系统产生生态毒性[10]。因此,有必要针对动物产品中残留的多肽类抗生素补充和确立更为准确高效的检测方法。然而,目前针对多肽类抗生素的检测大多只涉及到一种或两种化合物的检测方法研究,鲜有针对同时检测多种多肽类抗生素的方法研究,最多同时测定也不超过8 种[11-14]。主要有微生物法[15]、免疫分析法[16]、薄层色谱法[17-18]、毛细管电泳法[19]、毛细管电色谱[20]、高速逆流色谱法[21]、高效液相色谱法[22]和液质联用法[23-25]等。其中,液质联用法可以同时发挥色谱和质谱的优势进行定性、定量检测[26],非常适用于多肽抗生素的残留检测。本研究建立了一种UPLC-MS/MS 同时测定动物组织中9 种多肽类抗生素残留的方法,可为相关部门进行多肽类残留的标准制定和实际检测提供补充和参考。
1 材料与方法
1.1 材料与仪器
去甲万古霉素 纯度为83.4%,中国药品生物制品检定所;维吉尼亚霉素M1 纯度为90% 加拿大Toronto Research Chemicals 公司;万古霉素、粘杆菌素A、粘杆菌素B、杆菌肽A、太古霉素纯度分别为91.2%、84.7%、80.6%、47.1%、28.9% 德国Dr. Ehrenstorfer GmbH 公司;乙腈、甲醇、甲酸、正己烷 色谱纯,德国Merck 公司;盐酸、酸性氧化铝、C18、乙酸铵、草酸、三氯乙酸、磷酸 国药集团化学试剂有限公司;固相萃取柱Oasis HLB 美国Waters 公司,60 mg/3 mL;出口鸡肉、鸡肝、猪肉等待测样品共37 批 均采自福建省食品药品监督管理局日常抽检。
API5500 超高效液相色谱串联质谱仪 美国Applied Biosystems 公司;CR21N 高速冷冻离心机美国Beckman Coulter 公司;HN200 多功能氮吹仪 济南海能仪器股份有限公司。
1.2 实验方法
1.2.1 标准溶液的配制 根据9 种多肽类物质的纯度进行折算后,准确称取适量的9 种多肽类标准物质,先用0.1%甲酸水溶解并用甲醇定容至100 mL,分别配制成质量浓度为100 mg/L 的单标储备液,分别准确量取适量的上述标准储备液适量,用乙腈-0.1%甲酸溶液(20:80,v/v)配制成所需浓度的混合中间标准工作液,精密量取适量的中间标准工作液,分别用处理过的空白基质样品配制成不同质量浓度的系列标准工作液,现用现配。所有溶液于-4 ℃下冷藏避光储存。
1.2.2 样品前处理 取5 g 均质过的样品(精确到0.01 g)于50 mL 具塞塑料离心管,加入15 mL 甲醇+0.1 mol/L 盐酸溶液(7:3,v/v)和2~3 g 酸性氧化铝,振荡2 min,4000 r/min 下离心5 min。收集上清液,渣中再加入10 mL 甲醇-0.1mol/L 盐酸溶液(7:3,v/v)重复提取一次。合并上清液并定容至25 mL。取10 mL 于45 ℃氮吹干,用5 mL 0.1 mol/L 盐酸溶液复溶,再加入2.0 mL 正己烷,混匀,15000 r/min低温离心5 min,弃去正己烷,下层水溶液全部过玻璃纤维滤纸,待净化。
先活化Oasis HLB 固相萃取柱,依次加入3.0 mL甲醇、3.0 mL 水、3.0 mL 0.1 mol/L 盐酸溶液活化。将待净化液全部过柱,弃去流出液。用5.0 mL 0.1 mol/L盐酸溶液、3.0 mL 水淋洗,弃去流出液。最后加入2.0 mL 0.1%甲酸甲醇溶液(体积比)、3.0 mL 甲醇洗脱,收集洗脱液。洗脱液45 ℃下氮气吹干,用1.0 mL 乙腈-甲酸水(0.1%)(1:9,v/v)复溶,混匀后过0.22 μm 滤膜待测。
1.2.3 液相色谱条件 色谱柱:Luna® C8(2)100A(150 mm×2 mm,3 μm);柱温:35 ℃。流动相:A 为0.2%甲酸水溶液,B 为乙腈;梯度洗脱程序:1.5~3 min,10%B~75%B;3~5.5 min,75%B 不变;5.5~6 min,降至10%B,保持2 min,进样量:5 μL,流速:0.3 mL/min。
1.2.4 质谱条件 电离模式:电喷雾电离正离子模式;检测方式:多反应监测模式;离子源喷雾电压:5000 v;气帘气压力:0.275 MPa;离子源温度:500 ℃;雾化气压力:0.345 MPa;加热辅助气压力:0.345 MPa。
1.3 数据处理
本文实验重复三次,使用Microsoft Excel 软件进行数据分析处理及绘图。
2 结果与分析
2.1 检测条件的选择
2.1.1 质谱条件的优化 多肽类抗生素一般含有4~16 个氨基酸构成的线状或环状结构,分子量一般在300~3000 之间。在电喷雾离子化时容易与H+结合而使离子带正电,根据结合的H+数量不同,离子所带的电荷数可能是单个也可能是多个。实验表明,多肽类抗生素如万古霉素、去甲万古霉素、恩拉霉素、杆菌肽、太古霉素等在电喷雾离子化时能产生不同丰度的双电荷离子和三电荷离子。实验中发现前者的响应比后者要高,因此选择双电荷离子([M+2H]2+)分别作为母离子;粘杆菌素A、B 在流动相体系含有铵盐的情况下,带双电荷离子的响应和三电荷离子差异不大,而在流动性只有甲酸的情况下,带三电荷离子的响应比双电荷离子高大约100 倍,因此选择的三电荷离子([M+3H]3+)作为粘杆菌素A、B 的母离子;而维吉尼霉素M1 产生稳定的、强度较高的准分子离子峰([M+H]+)。因此,它被选作维吉尼亚霉素M1 的母离子。母离子断裂或重排之后会产生不同丰度的碎片离子。选择两个响应值高的离子作为定量和定性离子,再优化其他参数。采用多反应监测(MRM),使测定的灵敏度达到最大。9 种多肽类化合物的主要质谱分析条件参数见表1。
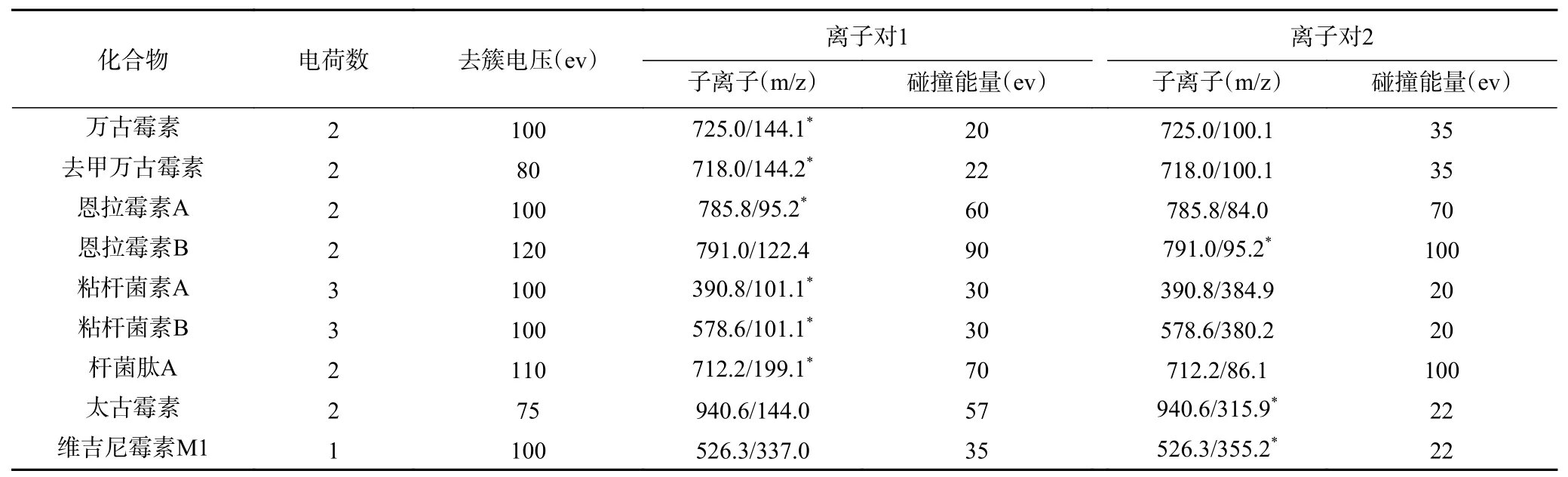
表1 9 种多肽类化合物的主要质谱分析条件参数Table 1 Main parameters for the 9 polypeptide antibiotics by mass spectrometry
2.1.2 色谱条件的优化 由于9 种化合物溶解性差异较大,通过调节流动相中缓冲溶液的pH、离子强度和有机溶剂(乙腈或甲醇)的含量,可以控制多肽类化合物的离子化强度和溶解性,在色谱柱上获得适当的保留和分离。流动相的组成和添加剂对分析物的峰形、基质效应和离子化效率均会造成一定的影响。本文在文献研究的基础上,考察了不同流动相组成对于分析物灵敏度的影响。常用的流动相有5 mmol/L 乙酸铵-乙腈、5 mmol/L 乙酸铵含0.2%甲酸水溶液-乙腈和0.2%甲酸水溶液-乙腈,本实验比较了这3 种流动相的效果。当所用色谱柱为反相色谱柱时,多肽抗生素易发生拖尾现象,原因在于其结构上含有多个氨基和羧基,而反相色谱柱中的硅醇基易和这些氨基羧基发生作用。当缓冲溶液为乙酸铵时,粘杆菌素、恩拉霉素和杆菌肽响应值较低,所以不选用5 mmol/L 乙酸铵-乙腈和5 mmol/L 乙酸铵含0.2%甲酸水溶液-乙腈;而加入甲酸后,响应值逐渐增高。因此选用0.2%甲酸水溶液-乙腈。分析其原因,可能是由于甲酸可以提供离子化的质子来源,所以加入甲酸能提高响应值。为了使9 种多肽类化合物在C8色谱柱上得到较好的分离效果,按照1.2.1 的条件进行梯度洗脱,多肽类化合物定量离子对的MRM 色谱图见图1。
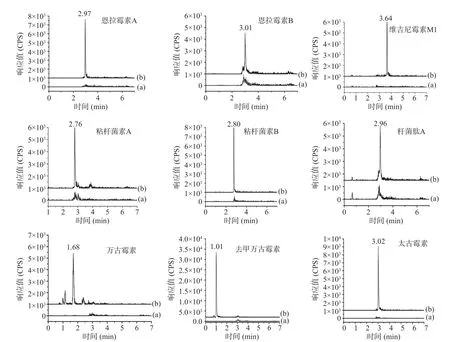
图1 空白基质(a)和鸡肉基质加标(b)的多肽类化合物定量离子对的MRM 色谱图Fig.1 MRM chromatograms of quantitative transitions of peptide compounds in blank matrix (a) and chicken matrix spiked (b)
2.2 样品前处理的优化
2.2.1 提取溶剂的选择 由于多肽类化合物的极性和溶解性差异较大,单一溶剂很难满足要求,为此实验考察提取溶剂种类、比例、酸性条件等对提取效果的影响。多肽类化合物属于极性化合物,其结构中包含多个氨基和羧基,宜采用极性有机溶剂提取,又因为本实验样品均为动物性食品样品,提取中必须有效去除脂肪沉淀蛋白[27-28],综合以上各因素,本实验首先比较甲醇、乙腈和乙酸乙酯的提取效果,结果表明,提取效果甲醇较好而乙酸乙酯最差,其中乙酸乙酯对去甲万古霉素、万古霉素、恩拉霉素的提取回收率均小于20%。随后为了比较甲醇在不同酸性体系下的提取效果,本实验选择甲酸、盐酸、草酸、三氯乙酸、磷酸来构建不同的酸性体系,结果(见表2)表明,甲醇中加入三氯乙酸、磷酸后提取回收率为40%~50%,加入甲酸、草酸后提取回收率为60%~70%,甲醇与盐酸结合的体系对9 种多肽类的提取效果更佳,提取回收率均在80%以上;再分别考察甲醇与盐酸不同比率(9:1;8:2;7:3;6:4;5:5,体积比)对化合物提取效果的影响,结果表明水相越高,目标物对去甲万古霉素、万古霉素的提取效果越好,而其他化合物的提取效果越差,考虑到提取溶液水相比率太高粘度增大,不利于后面的浓缩净化过程,因此综合考虑,选择甲醇-0.1 mol/L 盐酸溶液(7:3,V/V)作多肽抗生素的提取溶剂,各化合物的提取回收率均可达到85%以上。

表2 甲醇在不同酸性体系下对多肽类化合物的提取回收率Table 2 Extraction recoveries of peptide compounds with methanol in different acidic systems
2.2.2 提取条件的优化 动物组织样品提取液中杂质含量较多,这些杂质主要以蛋白、脂肪为主。为了尽可能的减少这些杂质对化合物的干扰而不影响提取回收率,实验在提取过程中加入一些净化填料。分别比较了加入酸性氧化铝、PSA 粉、C18粉、无水硫酸镁等对提取回收率的影响(见图2),其结果表明PSA 粉对粘杆菌素有明显的吸附作用;而C18粉也会影响太古霉素、杆菌肽、维吉尼霉素M1 的提取效果;无水硫酸镁对除去甲万古霉素和维吉尼霉素M1 外所有化合物的影响比酸性氧化铝的影响大;酸性氧化铝则对所有化合物的影响不大,故在前期提取时加入适量的酸性氧化铝去除蛋白等杂质[29-30],另外在过柱净化前加入正己烷除去脂肪类等杂质,可有效去除杂峰的干扰。
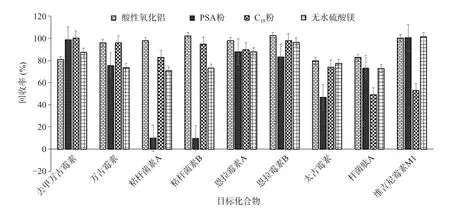
图2 不同净化填料对目标化合物回收率的影响Fig.2 Effect of different purification fillers on the recovery rate of target compounds
2.2.3 过柱净化方法的优化 根据参考文献[31-32],多肽抗生素样品净化方法主要采用阳离子交换、C18和HLB 固相萃取柱。由于化合物恩拉霉素在碱性溶液中不稳定,本实验重点比较了2 种不同的固相萃取柱Oasis HLB 和C18固相萃取柱,分别使用标准溶液进行固相萃取过柱实验,结果表明C18对去甲万古霉素和万古霉素的过柱效果较差,过柱回收率在20%以下,HLB 对几种抗生素的过柱回收效果较好,因此综合考虑选择HLB 固相萃取小柱净化样品。在使用HLB 小柱进行洗脱液的试验时发现粘杆菌素、杆菌肽会吸附在填料上,用纯甲醇无法将其洗脱下来,但是当向洗脱剂中加入适量酸的时候就可以洗脱,故实验采用分步洗脱的方式,先用含甲酸甲醇溶液(体积比1:100)洗脱杆菌素、杆菌肽等化合物,再用甲醇洗脱太古霉素、维吉尼霉素M1 等极性较弱的化合物,优化洗脱比例,其甲醇洗脱体积优化结果见图3,结果表明2 mL 0.1%甲酸甲醇溶液再加3 mL 甲醇洗脱效果最佳,9 种化合物的过柱回收率在92.1%~98.9%,适合定量分析。

图3 9 种多肽类化合物在HLB 上的洗脱结果Fig.3 Elution histogram of 9 polypeptide antibiotics on HLB
2.3 基质效应的考察
基质效应是开发和确证液相色谱串联质谱法过程中十分重要的一步。通常认为,基质匹配校正曲线斜率与标准曲线斜率的比值在0.85~1.15 之间时,基质效应可以忽略不计。实验以鸡肉、猪肝、猪肾为基体,以提取液配制基质曲线来评价不同的基质效应。结果表明,太古霉素(0.3)和粘杆菌素B(0.8)存在基体抑制效应,而其它化合物均在0.85~1.02 之间,不存在基质效应。综合考虑需消除基质效应的影响,采用基质匹配标准曲线定量。
2.4 方法学考察
2.4.1 方法的线性范围、检出限及定量限 按1.2.1的方法配制一系列质量浓度的混合标准工作溶液,在1.2.3 色谱条件和1.2.4 质谱条件下进行测定,结果表明在0.1~500 ng/mL 范围内,9 种分析物的质量浓度(x, ng/mL)与其定量离子峰面积(y)之间均有良好的线性关系,相关系数在0.9955~0.9999 之间(见表3)。方法的定量限(LOQ)根据其响应值大于或等于基线响应值的10 倍(S/N≥10)且准确度和精密度(以RSD 计)≤20%来确定。采用添加法进行实际样品的检测,得出维吉尼霉素M1 定量限为1.0 μg/kg,去甲万古霉素和万古霉素、恩拉霉素A、B、太古霉素的定量限为10 μg/kg,粘杆菌素A、B、杆菌肽A 的定量限为20 μg/kg。
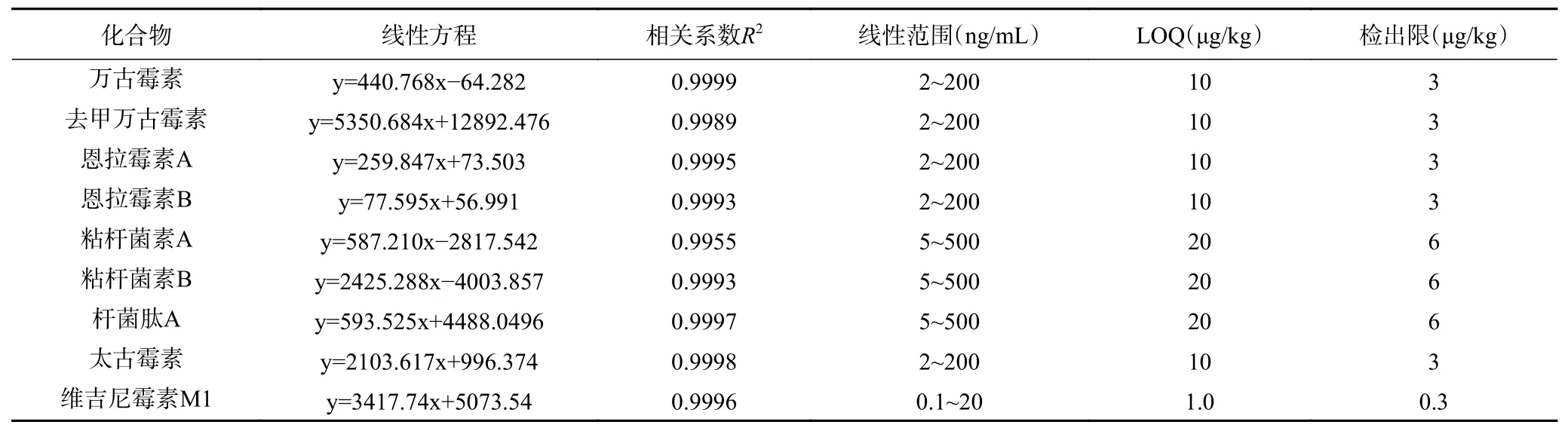
表3 9 种多肽类化合物的线性方程和相关系数Table 3 Linear equations and correlation coefficients of 9 polypeptide antibiotics
2.4.2 方法的回收率与精密度 取有代表性的不同空白样品(鸡肉、猪肉、猪肝、猪肾)作为添加回收实验的基质,进行添加回收和精密度实验。采用标准加入法[27],分别添加低、中、高3 个不同浓度水平的多肽抗生素混合标准溶液,每个添加浓度做6 组平行测定,计算平均加标回收率(Rce.%)及相对标准偏差(RSD%)。结果表明9 种多肽的平均回收率为74.2%~96.3%,精密度RSD 为4.3%~14.6%,表明本方法的准确性良好,精密度良好,结果见表4。
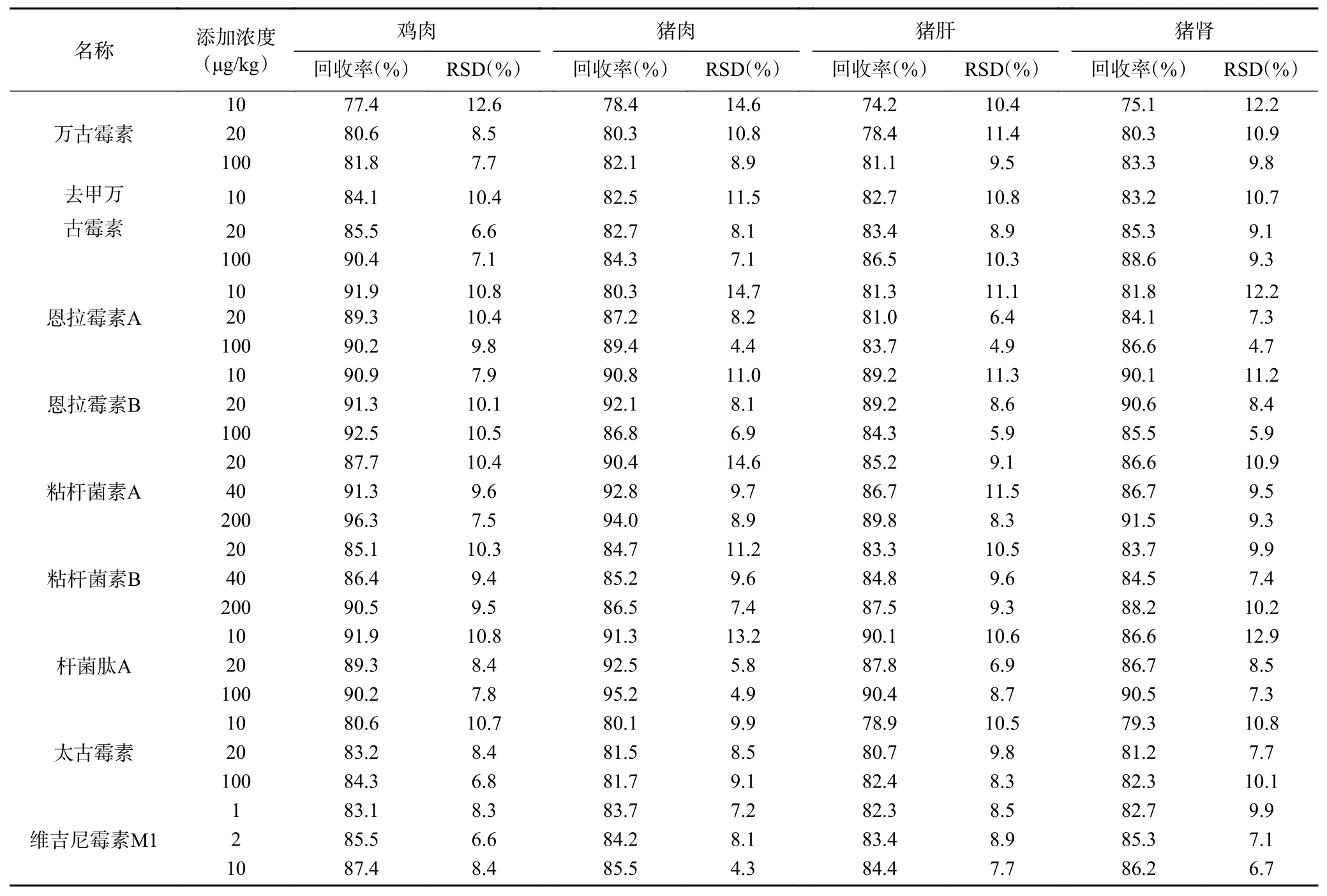
表4 9 种多肽类化合物在动物组织样品中的加标回收率和精密度(n=6)Table 4 Recovery and precision of 9 polypeptide antibiotics in animal tissue sample (n=6)
2.4.3 实际样品检测 应用本方法检测出口鸡肉、鸡肝、猪肉等样品共37 批,结果在2 批次鸡肝样品中检出粘杆菌素A,结果分别为26.3 μg/kg 和35.0 μg/kg,其他化合物均未检出。
3 结论
本实验采用超高效液相色谱串联质谱法对动物组织中的万古霉素、去甲万古霉素、恩拉霉素A 与B、太古霉素、粘杆菌素A 与B、杆菌肽A、维吉尼霉素M1 等9 种多肽抗生素残留进行了快速、准确的定量与定性分析,在鸡肉、猪肉、猪肝、猪肾等基质中采用加标回收试验验证方法的准确性,实验结果显示,本方法的回收率为74.2%~96.3%,相对标准偏差为4.3%~14.6%。采用所建立的方法对鸡肉、鸡肝、猪肉等样品进行检测,在鸡肝中检测到了粘杆菌素A。该方法简单快速,灵敏度高,可为有关部门进行动物组织中多肽类残留的标准研究和实际检测提供方法参考和补充。
