BCHE基因复合杂合变异致丁酰胆碱酯酶缺乏症临床特征和遗传分析
2022-08-26戴立英
王 娟, 余 静, 陈 珺, 郑 洪, 戴立英
(安徽省儿童医院新生儿科,安徽 合肥 230051)
丁酰胆碱酯酶(butyrylcholine eterase,BChE)和乙酰胆碱酯酶(acetylcholinesterase,AChE)是人类胆碱酯酶的2种类型,AChE主要分布于神经组织,BChE主要由肝脏产生并分布于所有组织[1]。BChE的生理功能尚不明确,但对含酯药物,包括某些短效神经肌肉阻断剂、普鲁卡因、丁卡因及海洛因等分解代谢具有重要的药理学意义[2-3]。丁酰胆碱酯酶缺乏症(butyrylcholinesterase deficiency,BCHED)患者通常没有生命危险,但由于其对肌肉松弛剂等药物敏感性较高,可能会在应用类似药物后长期昏迷,甚至死亡[4]。BCHE基因变异导致的BCHED(OMIM#617936)可引起人体血清胆碱酯酶异常降低,多数情况下无临床表现,但近年来偶有报道BCHED患儿存在智力障碍[4-5]。本研究对2例由BCHE基因复合杂合变异导致的BCHED患儿进行报道,并分析其遗传学特征。
1 材料和方法
1.1 研究对象
患儿1,女,汉族,出生26 d,系其母第2胎第2产,孕周39+4周剖宫娩出,Apgar评分为9~10分,患儿于出生20 d起无明显诱因出现“腹胀伴腹泻”,就诊于当地医院,黄色水样便,频次7~8次/d,给予地衣芽孢杆菌活菌胶囊和醒脾养儿颗粒治疗后,大便呈绿色伴奶瓣,频次较前稍减,但仍未能控制,后因“新生儿腹泻、新生儿肺炎”转院至安徽省儿童医院新生儿科。
患儿2,女,汉族,出生23 d,系其母第1胎第1产,孕周38+2周顺产,Apgar评分为9~10分,患儿因皮肤黄染半月就诊于安徽省儿童医院新生儿科。
2例患儿父母及患儿1的哥哥(3岁)身体健康,否认近亲结婚及家族遗传史。
1.2 方法
1.2.1 基因测序 患儿父母均签署知情同意书,经安徽省儿童医院医学伦理委员会批准,对患儿及父母行全外显子家系检测(trio-based wholeexome sequencing,trio-WES)。采集核心家系成员外周血各3 mL,乙二胺四乙酸抗凝。根据基因组提取试剂盒(北京康为世纪生物科技有限公司)说明书提取白细胞DNA,构建文库后用Illumina Miseq高通量测序仪(美国Illumina公司)对设计序列进行捕获,设计序列可覆盖基因全外显子区域及部分内含子区,参考序列为美国国家生物技术信息中心数据库人类基因组参考序列19版,利用GATK软件(美国Broad研究院)分析得出单核苷酸变异及插入、缺失等变异,过滤无效变异后,对可靠变异谱进行危害性预测分析。参考Ensembl数据库(http://grch37.ensembl.org)设计可疑变异引物,采用Sanger测序进行验证,检测仪器为ABI 3130XL分析仪(美国ThermoFisher Scientific公司)。
1.2.2 变异注释与蛋白结构模型预测分析 首先搜索公共数据库(SNP数据库、千人基因组数据库、ExAC数据库),统计变异位点在正常人群中的发生频率。在ClinVar数据库(http://www.ncbi.nlh.gov/clinvar/)和HGMD数据库(http://www.hgmd.cf.ac.uk/ac/index.php)中检索变异位点的报道情况。采用SIFT(http://provean.jcvi.org/index.php)、Polyphen-2(http://genetics.bwh.harvard.edu/pph2/)及Mutation Taster(https://www.mutationtaster.org/)在线平台分析变异位点的生物危害性,最后根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南[6]进行基因致病性分析。另外,本研究通过Uniprot数据库下载多物种BCHE蛋白序列文件,采用Mega软件(新西兰Mega公司)对变异位点进行同源性保守分析,采用Deepview软件(瑞士生物信息研究院)对错义变异进行模型分析。
2 结果
2.1 患儿临床资料
患儿1入院体格检查:神志清、反应可,全身皮肤无黄染。患儿2入院体格检查:面部及全身黄染,神志清、反应一般,皮肤弹性可。实验室检查提示2例患儿均出现胆碱酯酶异常降低,总胆红素(total bilirubin,TB)、直接胆红素(direct bilirubin,DBil)、间接胆红素(indirect bilirubin,IBil)升高。2例患儿丙氨酸氨基转移酶(alanine aminotransferase,ALT)、天门冬氨酸氨基转移酶(aspartate aminotransferase,AST)均正常,甲型肝炎病毒IgM抗体、丙型肝炎病毒抗体及乙型肝炎5项血清学指标均为阴性,粪便镜检未见真菌菌丝、真菌孢子、寄生虫卵,粪便轮状病毒及腺病毒检测均为阴性;入院前期未进行药物干预,且肝、胆、脾、胰超声检查未见明显占位性病变。因此,临床排除药源性胆碱酯酶降低。2例患儿各项指标检测结果见表1。
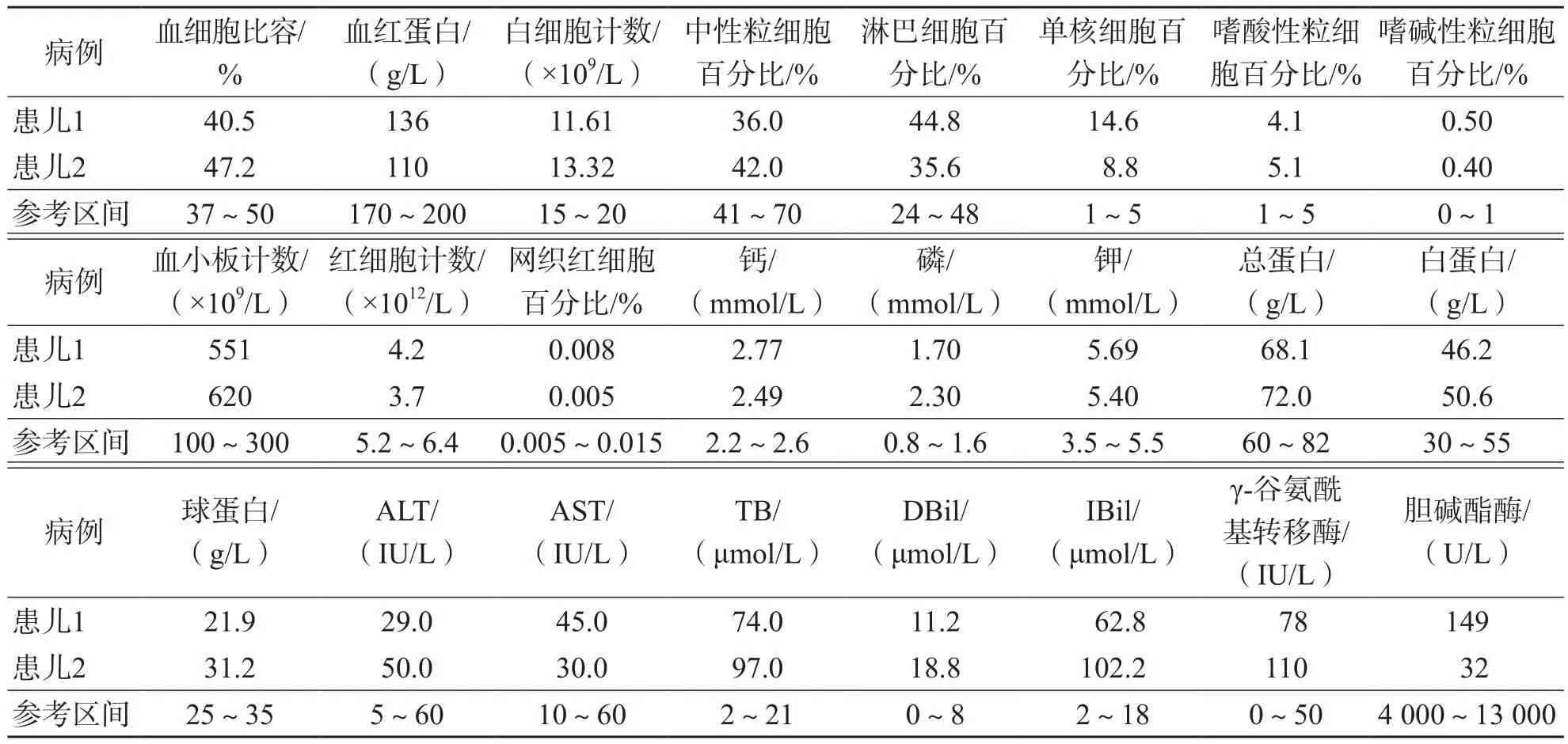
表1 2例患儿实验室检测数据
2.2 BCHE基因检测结果
2例患儿BCHE基因均发生复合杂合变异。患儿1为c.401_c.402insA/p.Asn134Lysfs*24和c.221delC/p.Pro74Hisfs*4,父亲携带杂合变异c.401_c.402insA/p.Asn134Lysfs*24,母亲携带杂合变异c.221delC/p.Pro74Hisfs*4。患儿2为c.401_c.402insA/p.Asn134Lysfs*24与c.127G>A/p.Gly43Arg,父亲携带杂合变异c.401_c.402insA/p.Asn134Lysfs*24,母亲携带杂合变异c.127G>A/p.Gly43Arg。Sanger测序证实存在变异位点。见图1。
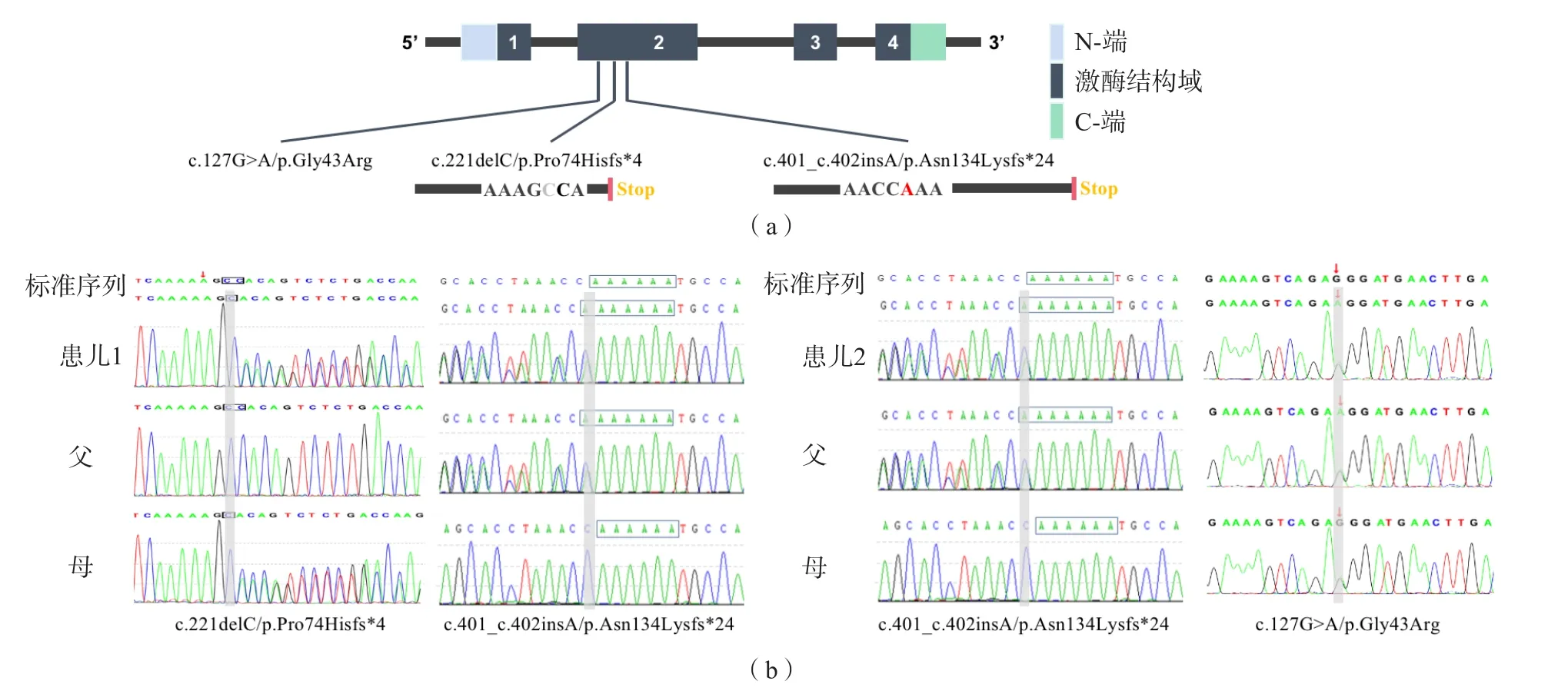
图1 BCHE基因检测结果
2.3 变异位点分析
对照正常人群变异分布,患儿1携带的变异位点c.401_c.402insA在Genome AD数据库中次要等位基因频率为0.000 9,另一个变异位点c.221delC在SNP数据库、千人基因组数据库及ExAC数据库中均未被收录,ClinVar及HGMD数据库也未见相关报道,属BCHE基因新的变异。患儿2携带错义变异c.127G>A,该变异位点经SIFT、Polyphen-2及MutationTaster在线分析,结果分别为0.009(有害的)、1.0(可能有害的)及1(致病的),预测该变异位点对蛋白功能可能存在危害性。依据ACMG指南,变异位点c.401_c.402insA/p.Asn134Lysfs*24为“致病性(PVS1+PS3)”、变异位点c.221delC/p.Pro74Hisfs*4为“可能致病性(PVS1+PM2)”,变异位点c.127G>A/p.Gly43Arg为“不确定性(PM1+PM2+PP3)”。
蛋白同源分析结果显示,p.Gly43在不同物种间高度保守,且位于BCHE蛋白活性区域。当发生p.Gly43Arg变异时,可导致其在空间结构上与p.Thr54残基发生碰撞,影响局部结构稳定性,进而可能产生危害性。见图2。
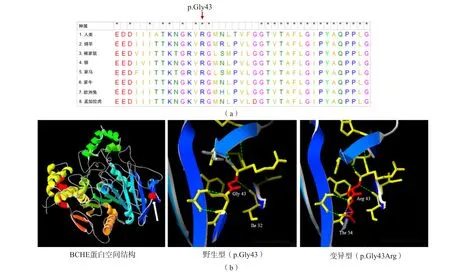
图2 变异蛋白空间结构分析
3 讨论
BChE是一种具有复杂分子结构的代谢酶,可对部分肌肉松弛剂(如琥珀酰胆碱)或普鲁卡因等进行高效水解[2,7]。缺乏BChE的患者因对部分药物代谢速度减缓,敏感性增强,较少出现疾病表型[5]。导致人体血清BChE降低的主要因素为肝脏疾病、肾脏疾病、营养不良、烧伤、体温过低和怀孕,次要因素为包括毒品在内的化学品及部分药物,如单胺氧化酶抑制剂[8-10]。BCHED是由BCHE基因变异导致的,目前至少已报道了70种以上的BCHE基因变异,此类出现遗传变异的患者通常无临床表现,但若在术前使用琥珀酰胆碱等药物,则易导致神经肌肉阻滞延长,进而引起呼吸暂停或昏迷[11]。本研究中的2例患儿均出现血清胆碱酯酶异常降低,其中患儿1首发症状为不明原因新生儿腹泻,而患儿2表现为新生儿黄疸,经实验室检测确认无肝功能损伤,且无药物中毒可能。BCHED患儿出现腹泻表现的发病机制虽尚不明确,但已有的研究结果或能解释其关联性。与AChE在调节胆碱能信号方面的明确机制不同,BChE生理功能至今仍不清楚[10]。在正常生理环境下,BChE既可水解乙酰胆碱,也可水解丁酰胆碱,而乙酰胆碱水平降低可阻断神经传导;当BChE严重缺乏时,乙酰胆碱水平会异常升高,并激活胆碱能受体,使得肌肉收缩、胃肠道功能紊乱、尿失禁、副交感神经张力增大,进而导致腹泻发生的风险升高[2,12]。
BCHE基因位于3号染色体q26.1-q26.2区间,由4个外显子组成,其编码的BChE呈中心富集脯氨酸的四聚体结构,可分布于全身组织,尤其高表达于肝脏、肺、大脑及心脏等器官,其表达量高于AChE,因此肝脏等器官损伤会引起BChE水平降低[13-15]。本研究2例患儿BCHE基因发生复合杂合变异,且均携带c.401_c.402insA杂合变异。有研究报道,1例具有轻度智力障碍的14岁中国男孩存在该变异位点,由于BChE与AChE共同调控神经节、轴突、神经末梢及椎体生长等生理过程,因此当BChE水平严重缺乏时,可能会下调相关神经系统功能,进而出现智力障碍的临床表现[5]。BCHE基因变异导致BCHED的报道较少,因此需更多样本以确定c.401_c.402insA杂合变异是否为我国人群的热点变异。另外,患儿1携带的c.221delC杂合变异和患儿2携带的c.127G>A杂合变异在GlinVar等数据库中尚未查到相关数据,属BCHE基因新变异,这也进一步拓展了BCHE基因变异谱-表型谱。
BCHED发病率在不同人群中有较大差别。BChE水平在正常水平10%以下的类型称为沉默型BCHED,在欧美人群中极为罕见,BCHE基因纯合变异发生率约为1/100 000[2];但在印度Vysya社区高频发生,有研究随机检测了该地区221名居民,有9例为BCHE基因纯合变异[15-16]。BCHED的另一种类型为K-型,BChE常低于正常水平的30%,该型在欧美人群及日本人群中最常见,约有1/70的患者BCHE蛋白第539号丙氨酸变异为苏氨酸(p.Ala539Thr),而在K-型纯合变异人群中至今尚未见肌肉松弛剂不良反应的报道[17]。我国尚无BCHED相关大规模队列研究,因此尚不清楚中国人群特异性变异类型及发病率。
综上所述,本研究分析了2例因BCHE基因复合杂合变异导致的BCHED患儿临床特征和遗传信息,发现2个BCHE基因新变异(c.221delC与c.127G>A)。c.221delC为功能丢失型变异,可能会导致蛋白功能降低;变异位点c.127G>A位于N-联糖基化区域,可能影响局部酶结合功能。BCHED通常无临床表型,但应用琥珀酰胆碱等肌肉松弛剂时会增加呼吸抑制及昏迷等风险,应避免对患儿使用这类药物。本研究患儿1出现新生儿腹泻症状是否与BCHE基因变异相关,仍需更多病例报道来进行综合分析,因此确定BCHE基因型与表型的关联性非常重要。
