A rare presentation of Guillain-Barre syndrome with GQ1b positivity: A case report
2022-08-26MinuGeorgeNeenaBabyPradeepMathewKoshyRajendranUllatilSureshkumarRadhakrishnan
Minu George, Neena Baby, Pradeep Mathew Koshy, Rajendran Ullatil, Sureshkumar Radhakrishnan
1Department of Neurology, Renai Medicity Multi Super Speciality Hospital, Kochi, Kerala, India
2Department of Nephrology, Renai Medicity Multi Super Speciality Hospital, Kochi, Kerala, India 3Department of General Medicine, Renai Medicity Multi Super Speciality Hospital, Kochi, Kerala, India
ABSTRACT
KEYWORDS: ChAdOx1nCoV-19; Papilledema; Albuminocytological dissociation; Guillain-Barre syndrome; Acute inflammatory demyelinating polyneuropathy
1. Introduction
Guillain-Barre syndrome (GBS) is a monophasic acute polyradiculoneuropathy which is autoimmune in nature, presenting with rapidly ascending weakness with areflexia, reaching its nadir in less than 4 weeks. Clinical presentations such as headache, vomiting and papilledema are rare with GBS. Here we report a case of acute inflammatory demyelinating polyneuropathy (AIDP) following ChAdOx1 nCoV-19 vaccination, who presented with these features with GQ1b positivity.
2. Case report
Informed consent was obtained from the patient for the publication of this case report.
A 35-year-old female, with a history of gestational diabetes and pregnancy induced hypertension, presented with headache and vomiting 68 days postpartum. The patient took first dose of ChAdOx1 nCoV-19 vaccination 13 days prior to her present symptoms. Later she developed dysphagia, dysarthria and weakness of bilateral upper limbs with gait difficulty. She had also mild joint pain for which she took treatment from a rheumatologist. There was no associated fever, gastroenteritis, respiratory tract infection or any alteration of sensorium, double vision or drooping of eye lids.
The patient was conscious, and fundus showed bilateral papilledema.Gag reflex was sluggish with bifacial weakness. Motor examination showed neck muscle weakness with upper limb power of grade 3/5 and lower limb power of 4/5 as per the Medical Research Council grading and deep tendon reflexes were sluggish to absent. Sensory examination was normal and there were no cerebellar signs. Her routine blood investigations were within normal limits. Anti-DS DNA was negative (5.4 IU; reference range <100 IU). Rheumatoid factor(<10 IU/mL; reference range <14 IU/mL), anti-cyclic citrullinated peptide antibody (8 RU/mL; reference range 0.2-8.0 RU/mL)and complement levels (complement C3 and C4, 125, 29 mg/dL,respectively; reference range 90-180 and 10-40 mg/dL, respectively)were within normal limits. Magnetic resonance imaging of brain with magnetic resonance venogram showed flattened posterior sclera with prominent subarachnoid space around optic nerves with stenosis of bilateral transverse sinus with possibility of intracranial hypertension. Hence lumbar puncture was done with pressure monitoring, showed cerebrospinal fluid (CSF) opening pressure of 32 cm of water with evidence of albuminocytological dissociation(protein of 112.3 g/dL, no cells) with CSF sugar of 87 mg/dL (blood sugars were 120 mg/dL). In view of the above findings, diagnosis of AIDP was considered. SARS-CoV-2 antibody titer was not conducted as the patient’s condition improved and her relatives were not willing for the same. Nerve conduction study (NCS) on the fifth day after onset of symptoms showed severe compressive neuropathy of right median nerve which was not contributory. Five cycles of plasmapheresis were used and physiotherapy was initiated. NCS was repeated on the ninth day of illness and showed the same findings.Antiganglioside antibody panel done showed GQ1b IgG antibody positivity. NCS repeated after two weeks showed conduction block from left peroneal nerve, prolonged distal latencies from bilateral median, reduced compound muscle action potential amplitude from right median nerve (NCS findings are shown in Table 1). Sensory nerve action potential (SNAP) was absent from bilateral median and ulnar nerves with preserved sural SNAP. The condition of patient improved gradually and her palatal and neck muscle weakness improved. She could walk without support and was discharged after 20 days of admission.
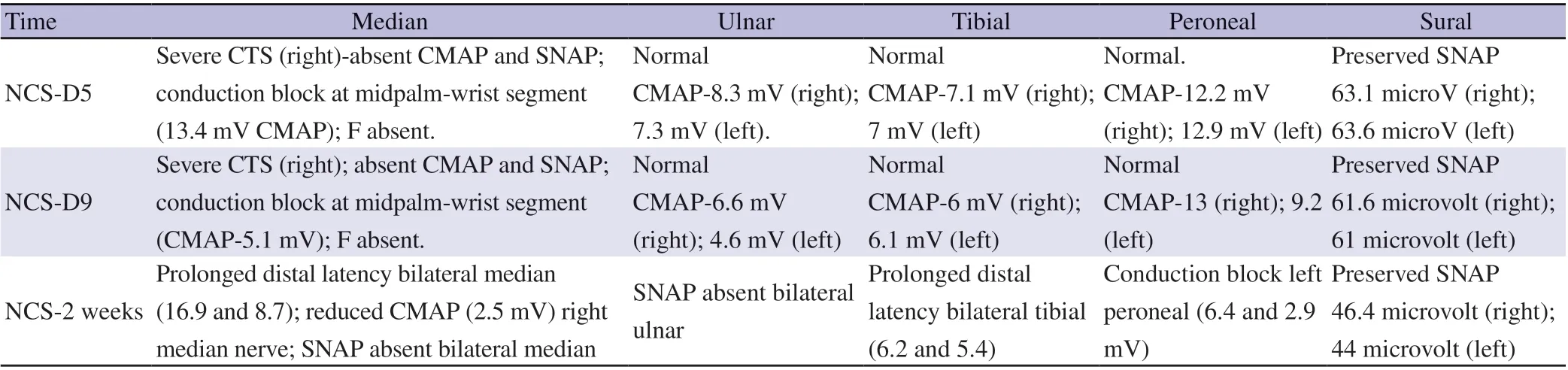
Table 1. Nerve conduction study on day 5, day 9 and 2 weeks of a 35-year-old female Guillain-Barre syndrome patient with GQ1b positivity.
3. Discussion
The diagnostic possibilities considered in our case were: 1) Cerebral sinus venous thrombosis in view of initial presentation of headache,vomiting and bilateral papilledema. 2) Brain stem encephalitis in view of brain stem features like dysphagia and dysarthria. 3) AIDP atypical variant in view of proximal weakness and sluggish to absent reflexes. Albuminocytological dissociation and GQ1b positivity confirmed the diagnosis of AIDP.The pharyngeal-cervico-brachial, a rare (3%) variant of GBS,presents with ptosis, facial, pharyngeal and neck flexor muscle weakness that spreads to the arms and spares leg strength, sensation and reflexes mimicking botulism[1].
Albuminocytological dissociation with elevated protein in the absence of white blood cell elevation (less than 20 per mm3) is seen in 90% of cases. High protein level can cause obstruction of absorption of CSF in arachnoid villae and consequently raised CSF pressure[2]. High CSF pressure causes rise of pressure in the optic nerve sheath, which produces axoplasmic flow stasis in the surface nerve fiber layer and prelaminar region of the optic nerve head resulting in papilledema. High protein levels thus can result in elevated CSF pressure and papilledema[3] as well can cause compression of exiting nerve roots that may explain the proximal muscle weakness seen in some cases.
The earliest findings in AIDP are prolonged F-wave latencies and F-wave impersistence, due to demyelination of nerve roots. Absent SNAP in upper limb with normal sural nerve amplitude is highly diagnostic of AIDP. This is followed by prolonged distal latencies(due to distal demyelination) and temporal dispersion or conduction block. However, the sensitivity of NCS based on reported criteria may be as low as 22% in early AIDP[4], rising to 87% at five weeks after onset of the illness[5].
Antiganglioside antibodies play an important role in the immunopathogenesis of GBS. One of the proposed mechanisms is molecular mimicry, in which antecedent infection produces specific antibodies, which in turn cross reacts with peripheral nerve components because of the sharing of cross reactive epitopes[6].These antibodies can be directed against myelin or the axon.GQ1b antibodies are highly sensitive and specific to Miller Fisher syndrome but can also be seen in pharyngo-cervico-brachial variant,pure sensory ataxic variant and Bickerstaff encephalitis. The presence of GM1 ganglioside antibodies in Acute Motor Axonal Neuropathy and Acute Motor Sensory Axonal Neuropathy variants were associated with bad prognosis.
Plasma exchange involves removing subject’s plasma and replacing it with fresh frozen plasma or plasma substitute. Plasma exchange removes humoral factors such as autoantibodies, immune complexes,complement, cytokines and other nonspecific inflammatory mediators and was the first treatment shown in randomized controlled trials to be effective in GBS[7,8].
Two case series have been reported from South India, in which patients developed GBS following ChAdOx1 nCoV-19 vaccination[9,10]. In both reports, presentation of clinical symptoms happened within two weeks of vaccination. In our case,symptoms started on the 13th day of vaccination. All patients in the case series presented with facial diplegia and areflexic flaccid quadriparesis. In contrast, our patient presented with pharyngocervico-brachial variant with papilledema with relatively normal NCS findings compared to other patients in the case series[9,10].Albuminocytological dissociation and GQ1b positivity confirmed the diagnosis.
Atypical features in our case include initial presentation with headache, vomiting and papilledema which suggest an intracranial pathology, rostro-caudal progression of illness which suggest a cervicobrachial variant, evidence of albuminocytological dissociation with normal initial nerve conduction results and GQ1b positivity.
Though a temporal association between vaccination and development of present symptoms has been postulated, the exact aetiology of illness is difficult to document in the present scenario.The possibility of other infections are not completely ruled out.
4. Conclusions
Rare presentations like cervicobrachial variant and other variants can happen in AIDP. Strong index of suspicion is necessary so that rare presentations of AIDP are not misdiagnosed and early treatment can be instituted. In case of any suspicion specific antibody titers has to be checked to confirm the diagnosis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Funding
The authors received no extramural funding for the study.
Authors'contributions
MG and NB involved in concept, design, literature search, data acqusition, data analysis and manuscript preparation. PMK is involved in data analysis and manuscript preparation. RU and SR did critical review and supervised the project. MG acted as guarantor of manuscript.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Hurdles in achieving the goal of malaria elimination by India
- Conventional treatments and non-PEGylated liposome encapsulated doxorubicin for visceral leishmaniasis: A scoping review
- Prevalence and factors associated with belief in COVID-19 vaccine efficacy in Indonesia: A cross-sectional study
- Mosquito larva distribution and natural Wolbachia infection in campus areas of Nakhon Ratchasima, Thailand
- Genetic variation of sand flies (Diptera: Psychodidae) in Gampaha and Kurunegala districts of Sri Lanka: Complementing the morphological identification