Conventional treatments and non-PEGylated liposome encapsulated doxorubicin for visceral leishmaniasis: A scoping review
2022-08-26SoumyaRanjanSatapathyRudraNarayanSahoo
Soumya Ranjan Satapathy, Rudra Narayan Sahoo
1School of Pharmaceutical Sciences, Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar, India 2School of Pharmacy and Life Sciences, Centurion University of Technology and Management, Odisha, India
ABSTRACT Visceral leishmaniasis (VL), also known as Kala-azar, is caused by Leishmania (L.) donovani complex, which includes L. donovani and L. infantum and is associated with a high death rate as compared to the cutaneous and subcutaneous form. Treatment of VL includes chemotherapeutic agents which are associated with some major hurdles like toxicities, parenteral administration, high cost, parasite resistance and stability. Hence, there is an urgent requirement to develop novel chemotherapeutic agents or repurposing of existing drugs against VL. Developing formulation of new chemical entity for the treatment of VL is laborious, time consuming and associated with huge financial burden. However, screening of existing chemotherapeutic agents is a good alternative to avail cost-effective treatment option for VL. Non-PEGylated liposome encapsulated doxorubicin (Myocet®) is proposed as an alternative treatment option for VL in this review article. Here, we covered the fundamental aspects of VL, loophole associated with available current treatment strategies and non-PEGylated liposome encapsulated doxorubicin as a novel alternative formulation for treating VL, as this liposomal delivery system of doxorubicin might passively target the intracellular regions of macrophage.
KEYWORDS: Visceral leishmaniasis; Doxorubicin; Passive targeting; Repositioning; Non-PEGylated liposome encapsulated doxorubicin; Resistance
1. Introduction
Leishmaniasis is a poverty associated and derelict tropical microbial disease. Infected macrophages associated with reticuloendothelial system are mainly responsible for causing this infectious leishmaniasis disease. Worldwide, it has been considered as a third most prevalent vector-borne infectious disease after malaria and lymphatic filariasis in developing countries[1]. The parasite of visceral leishmaniasis (VL) was named by its co-investigators(William Leishman and Charles Donovan) as Leishmania (L.)donovani. VL is rife in 89 countries, wherein it is affecting a population of 350 million, posing at high menace of contagion. It is projected that 12 million people have been infected. Annually,there is an assessment explicating that 2 million leishmaniasis cases were reported. Among them, 1.5 million cases are of VL and half a million cases are of cutaneous leishmaniasis (CL)[2-5]. VL is a devastating, chronic and deadly disease with highest death rate compared to cutaneous and subcutaneous form of leishmaniasis.The mortality is as high as 100% within 2 years if left untreated as per WHO. It is regarded as kala-azar in India. Causative organism of fatal VL is L. donovani complex, which includes L. donovani and L. infantum. Previously, sand fly genus Phlebotomus is the vector and now Leutzomine is the vector for transmitting VL as systemic parasitic disease. The Leishmania species is zoonotic and anthroponotic in nature. Parasites showing bio-genetic organisms commute between a flagellated promastigote in the gut of the sand fly and an intra-cellular amastigote in the mammalian host. VL parasite drifts to central organs (liver and spleen) and replicates in macrophages until the host cell explodes. VL is characterized by symptoms such as persistent bouts of high fever, cough, stomach pain, weight loss,epistaxis, progressive enlargement of liver and spleen, cachexia,pancytopenia and sometimes bronchitis and diarrhea. VL is a most guileful infection usually occurring simultaneously with human immunodeficiency virus (HIV) infection as well as human migration and resettlement are altering the epidemiology and create hurdles for diagnosis and case management[6,7]. Sometimes, VL infected patients gradually suffer from illness and die, if left untreated.
2. Ontogenesis and morphlogy of Leishmania parasite
L. donovani is a digenetic parasite persisting in two hosts during its ontogenesis, i.e. man as the primary host and sand fly as the secondary host. Morphologically, it persists in both shapes as amastigote (leishmanial) and promastigote (leptomand)[1,2,8].
Morphologically, amastigotes are non-flagellate and are being as oval shaped (2-5 μm in length; 1-2.5 μm in width) with a central nucleus (<1 μm in diameter). Perpendicular to the nucleus, a minute structure lies called kinetoplast with DNA and mitochondria where as a basal body with axoneme represents the root of the flagellum. The amastigote divides and multiplies inside reticuloendothelial system of human by binary fission, and it ruptures above 50-200 cause. The liberated parasite enters blood circulation and invades other healthy cells of reticuloendothelial system[8,9].
Amastigote transforms into promastigote when female sand fly sucks blood of reservoir host. These changes occur when parasite enters into the gut of sand fly. Promastigotes are long, slender,spindle and flagellar in nature (with 15-20 μm length, 1-2 μm width).Morphologically, it is nucleated centrally with a basal body and kinetoplast, which is present near the anterior end. A single long flagellum originates beside the axoneme, which is almost equivalent to size of the body or longer than its body size. It undergoes reproduction asexually by binary fission process. By this fission procedure, promastigotes move towards pharynx, buccal cavity and proboscis of insect host. When sand fly takes plant juice, after the first infected blood meal, it multiplies and when such parasite infected sand fly bites a healthy person, the new-fangled host shows infection. Promastigotes penetrate and invade the reticuloendothelial system of new host and transforms into amastigote form to complete its cycle (Figure 1)[10,11].
Several chemotherapeutic agents have already been available in the market for treatment of VL, however, they are associated with a few limitations including frequent administration, cost, toxicities,parasite resistance and stability[2,12,13]. Penta-valent antimonials are the most favorable leishmanicidal agents, however, they are not preferred owing to their cardio-toxicity, hepatic and renal insufficiency. Pentamidine and amphotericin B are utilized as second line agents, but toxicity, availability and majorly cost limits their use[14,15]. Thus, there is requirement and development of new drugs and therapy along-with these common chemotherapeutic agents in the therapy of VL complex disease. Due to the unavailability of vaccine, many drugs are used to treat VL, as a result, leishmanial parasite develops resistance against drugs. Hence, aiming to reduce multiple drug resistance (MDR) in protozoal research, novel target identification and development of new drugs are the two main focusing points. Development of dual drugs or multiple drugs combination therapy should be aimed to avoid the drug MDR developed by parasite. Combination therapy is used for the safety and optimization of component drugs, which are administered either concomitantly or subsequently. In combination therapy, shorter regimen is more affordable, which is easier to administer and gives better adherence than current longer regimen. It also protects the life span of individual drugs[16]. An urgent need is persisted to establish a robust, storage-stable and affordable drug delivery system for new therapeutic agents particularly for this group of disease.
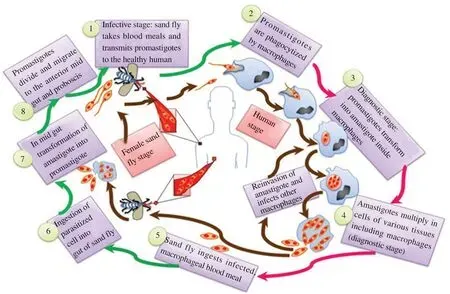
Figure 1. Life cycle of Leishmania parasite.
In the early 1990s, the potency of doxorubicin (Dox) was discovered for VL. Several investigators studied and found the equal effectiveness of Dox (in comparison to gold standard drug;amphotericin B) as an anti-VL agent. It was reported that Dox,an anti-cancerous agent, has potent inhibitory action against VL, but its clinical use is restricted due to the their several toxic responses[17-20]. In this review, we summarized the general facts and promises, loopholes associated with current treatment strategies and non-PEGylated liposome encapsulated doxorubicin (Myocet®) as a novel formulation for VL. Several research studies have considered liposomal Dox to be utilized in passively targeting the intra-cellular regions of macrophage with improved therapeutic outcomes as well as minimal toxicities as a new approach for VL treatment.
3. Conventional treatment
Development of resistance against pentamonial antimonial is an alarming issue as well as a major cause of their therapeutic discontinuation. Amphotericin B encapsulated colloidal and lipidic vesicle formulations are current choice for effective treatment of VL,however, excessive use of these formulations could lead to parasite resistance in the near future. Various chemotherapeutic agents available for the treatment of VL have been summarized in Table 1.
Since mid-1990s, pentavalent antimonial has been the foremost recommended drug for the therapy of VL and CL. Later, this situation was changed. From the last few decades, other drugs are used alternatively. Not only that many novel formulations of standard drugs came into existence and get registered for therapeutic use to treat VL, but also several drugs are in clinical trial for both diseases(VL and CL) and all these are summarized in Table 2. Currently used chemotherapeutic drugs or intended chemotherapeutic drugs to the treatment of VL are discussed below in brief.
3.1. Pentavalent antimonial
Pentavalent antimonial together with sodium stilbogluconate (SSG)and meglumine antimonite (MA) are the first line and standard drugs used in treating VL[44]. Antimonials acts as enzyme inhibitors.They act by blocking the enzymes that are regulating the oxidation of glucose (glycolysis and citric acid acycle) and β-fatty acids.Glycolysis and citric acid cycle are majorly responsible for adenosine triphosphate (ATP) production in Leishmania. In glycolysis and kreb's cycle, adenosine diphosphate undergoes phosphorylation by acquiring an inorganic phosphate and forms adenosine triphosphate in the presence of nicotinamide adenine dinucleotide (NADH)enzymes. Hence antimonials block the oxidation pathways for glucose and decrease ATP level, which is an important source for the existence of Leishmania[45]. Antimonials interfere the Leishmania amastigotes bioenergetics process. Numerically, clinically and economically, unresponsiveness of antimonials against VL is a serious problem. Chemotherapy of VL through SSG started with a dose of 10 mg/kg in the initial 6-10 days and subsequently increased unresponsiveness in India led to successive upward revisions in the dose. Presently, the dose of the drug is 10 times more in comparison with the other two earlier. There are lots of clinical reports showing serious issues including emergence of resistance and over 60% treatment failures in Bihar in India render the drug useless for routine use[46]. Various studies performed by experts using antimonials are summarized in Table 3. This data also explains the enhancement of dose over a period of time. Moreover, antimonials already have several other restrictions such as requirement of 3-4 weeks of parenteral therapy along with toxic effects like arthralgia,nausea, pancreatitis and abdominal pain and long term use shows cardio-toxicity[47]. Antimonial marketed products are expensive and batch to batch variability is also reported. Relapses after inadequate treatment, resistivity is also a limitation due to wide misuse of the drugs, inappropriate use by unqualified medical practitioners and easy availability of over the counter drugs[44]. All these facts collectively make antimonials useless for the treatment of VL. It is a serious alarming situation and therapies with alternate drugs become essential.
3.2. Pentamidine isothionate
Pentamidine (isothionate or methane sulphonate), an aromatic analogue of diamidine that is employed as a second line drug in the management of VL and was tested to be beneficial in SSG resistant VL cases in India[53]. High cost, irreversible toxicities,diabetes mellitus and mortality were the limiting factors for the use of pentamidine as the therapeutic agent[54]. A rapid decrease in the response rate from more than 95% cure rate in the early 1980s to less than 70% a decade later has led to the total abandonment of this drug in India[35]. Pentamidine cellular target is still not known,however, it acts on mitochondrial level, hampering the replication and transcription of pathogen.

35%-95%(depending ongeographic area)[22]Lengthof treatment Painful injection Toxicity Resistance Availability Quality control Highcost Frequentfatigue[6]Nausea Muscleandjointpain Increased transaminase Changes in ECG[19]Cardiotoxicity[19]Hepatic andrenal dysfunction[20]Pancreatitis[20]Shock andsudden death[21]0%allregions 5%in South Asia(doseof≥ 10 mg/kg)90% in other regions (dose of20-30mg/kg) [22,27]Low IV infusion Limited dose Heatstability Highprice Efficacy Optimum dose need to be defined Administrationin hospital Current alternative second-line treatment[21,22]Nephrotoxicity[23,24]Hypokalemia[25]igors[26]Fever[6,21,22]90% in South Asianotclearly established inother regions[27]Highprice Teratogenicity[31]Patient compliance Potential for resistance Nephrotoxicity[29]Hepatotoxicity[29]Teratogenicity[30]Gastrointestinaleffects(in 20%-55%patient,mildeffect)[31]Diminished biologicalreduction ofantimony (Ⅴ)to antimony(Ⅲ)[17]Reduced accumulation Gene amplification[18]60% treatment failure 39.7% treatment failure Not documented Not documented Not documented Not documented Possibility of parasitic resistance Reduced accumulationof miltefosine[28]Laboratory isolates 30days(dependingon geographical area)IMor IV 20mg/kg/day Pentostam®(GlaxoSmithKline)Sodium stibogluconate 30days(dependingon geographical area)IMor IV 20mg/kg/day Glucantim(Aventis)Meglumine antimonite Antimonial drugs 20days(dependingon geographical area)7-20 mg/kg/day SlowIV Fungizone(Bristol-Myers-Squibb,USA)AmphotericinB Deoxycholate 5-10doses over 10days over5days 10-20 mg/kg/day SlowIV 10-15 mg/kg/day SlowIV AmBisomeTM(Gilead Nexstar)Amphocil/ Amphotec(liposome technology,Sequesand Zeneca)Liposomal AmphotericinB Amphotericin B colloidal dispersion Polyene Antibiotic 10-15 mg/kg/day SlowIV over 10 days Abelcet(Liposome Co)Lipid complex over28 days Oral 1.5-2.5 mg/kg/day ImpavidoTM(Paladin)Miltefosine Alkyl phospholipid

Table 1. Continued.
3.3. Amphotericin-B (AmB)
From the genus Streptomyces bacterium, AmB was produced and isolated. Chemically, it is a polyene antibiotic generally employed for treating harmful fungal infections. This antifungal agent is amphiphilic in nature and employed as an alternative drug and second line drug in the therapy of VL after antimonial. Thus, it acts as an efficient anti-leishmanial agent but still it has certain cons such as high cost, limited availability, prolonged duration of therapy (up to 30 days), toxicity like infusion related side effects (fever, chills,bone pain, thrombophlebitis), hypokalemia, renal impairments and anemia. Apart from these side effects, it cured more than 97% of the leishamaniasis[55,56].
Anti-leishmanial activities of AmB are as follows: pharmacological action of AmB is to bind with the ergoesterol, a main component of leishmanial parasite and cause cell leakage of parasite thereby parasite rapidly loses many important monovalent ions. Despite the fact that in lesser degree, AmB reacts with cholesterol of host macrophages and inhibits the binding of parasites to cell. At higher concentration, it induces aqueous pore formation in cellular membrane leading to parasitic death[57-62].
Lipid-based formulations of AmB are more efficacious and well tolerated in comparison to usual preparations because their outstanding and unique physiochemical properties favor more uptake in targets of VL, i.e. RES (liver, spleen and bone-marrow etc.).United State of Food and Drug Administration (USFDA) approved the Ambisome®(unilamellar liposomal formulation), Amphocil®(colloidal dispersion) and Abelcet®(lipid complex) as some of the lipid-based formulations indicating for the therapeutic use against VL[63]. Amoung them, AmBisome®and Amphocil®were found more effective than Abelcet®formulation[64-66].
3.4. Miltefosine
Miltefosine (hexadecyle phosphocholine) is a membrane activating alkyl phospholipid, initially developed as an anticancer drug[67].It is a highly effective and orally active anti-leishmanial agent.In mid-1980s, its anti-leishmanial activity was discovered and ImpavidoTM, Profounda, Inc., Orlando, United States was the first approved commercial product for the miltefosine treatment of VL.Dose dependent studies showed 95% cure rate of 100 mg/day daily dose for 4 weeks in adults and 2.5 mg/kg in children[38]. Table 4 summarized the clinical trials data using miltefosine. Miltefosine is well tolerated orally without the need of close monitoring and also suitable for ambulatory treatments due to its high efficiency, low cost and short course of treatment as well as reduced side effects.Teratogenicity and abortificient activity are the two drawbacks associated with miltefosine, not permitting it to be used duringpregnancy[29]. It was reported that miltefosine showed gonadal toxicity (swollen testicles) in male dogs. Longer half-life (7 days)and low therapeutic index of drugs expand the chances of emerging resistance. Therefore, combination chemotherapy would be an alternative to avoid the development of resistance.
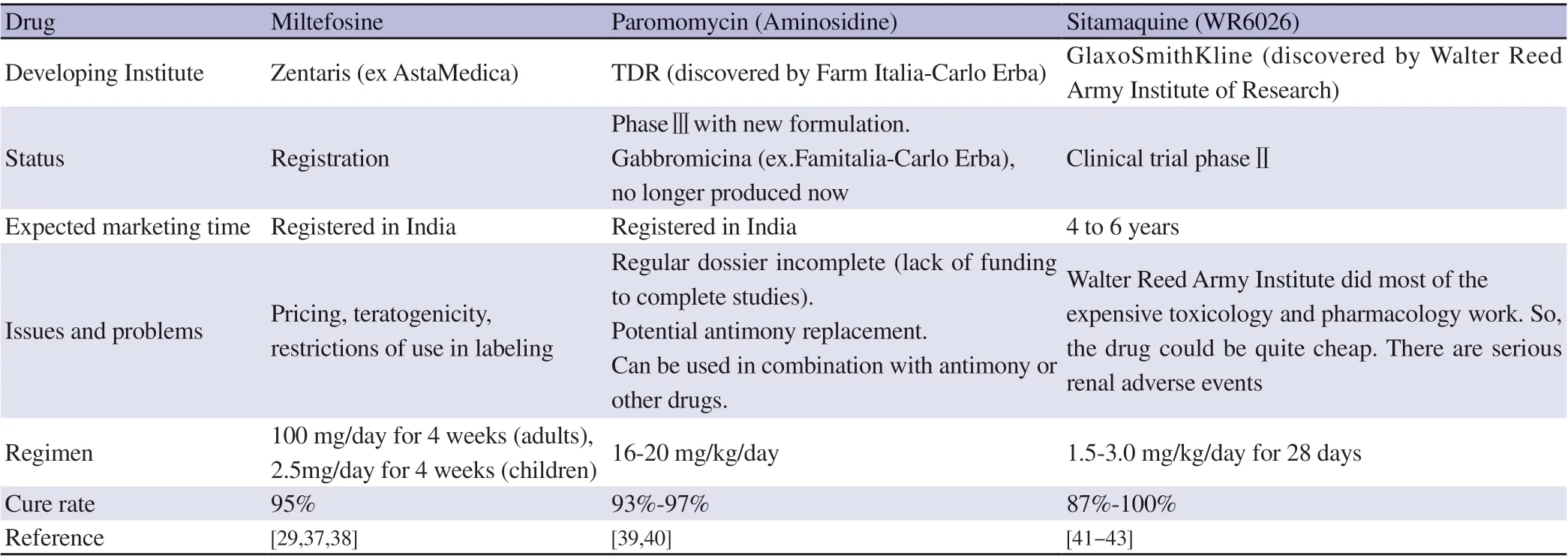
Table 2. Drugs under research and development pipeline.
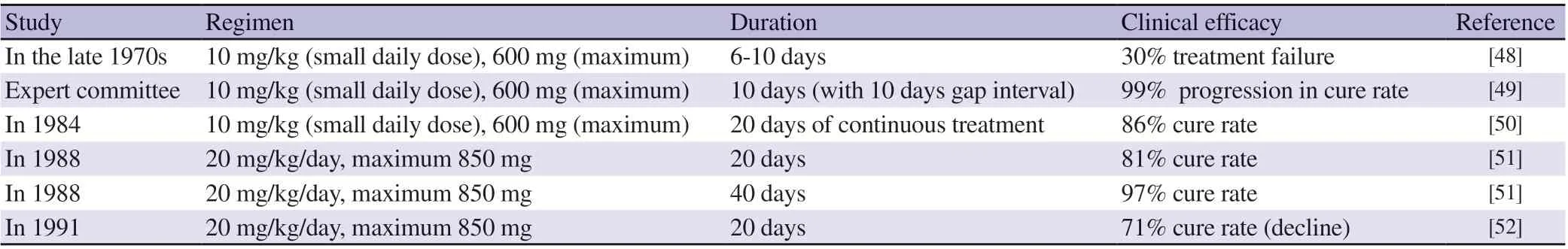
Table 3. Summary of antimonials employed studies.
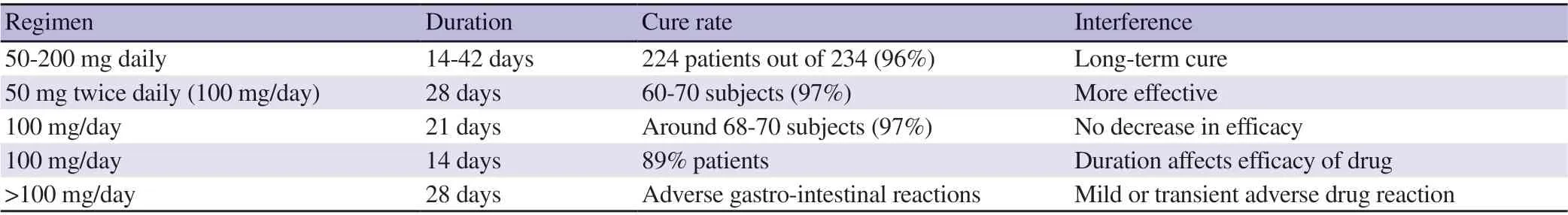
Table 4. Clinical trial studies of miltefosine[68,69].
3.5. Paromomycin
From the microbial culture of Streptomyces rimosus, paromomycin(aminosidine) antibiotic was produced and isolated. Chemically,paromomycin is an aminoglycoside antibiotic which was identified as an antileishmanial compound in the 1960s and can be employed to treat both VL and CL[33]. It has poor oral absorption, therefore, it is beneficial in the development of parenteral formulation. In Bihar,India, the phase Ⅲ clinical trials in Table 5 conducted revealed that paromomycin has high efficacy and safety (not inferior to AmB)because treated patients didn’t show nephrotoxicity and only a small percentage of inner ear damage with an increase in hepatic transaminases. Furthermore, paromomycin is active against different pathogens including bacteria. It has short duration of administration and low cost compared to other available drugs. It acts by promoting ribosomal sub-unit association of both cytoplasmic and mitochondrial forms, due to the dissociation caused by low Mg2+concentration and it also induces respiratory dysfunctioning of leishmania promastigotes[70-72].

Table 5. Aminosidine and sodium stilbogluconate studies in Bihar*[40].
3.6. Sitamaquine
Sitamaquine (8-aminoquinoline; a primaquine derivative)antimalarial drug was developed by Walter Reed Army Institute,Maryland, USA in collaboration with Glaxo Smith Kline,Philadelphia, USA for VL. For the past 8 years, it was still in the development stage and presently it is in clinical trial phaseⅡfor the therapy of VL[73]. It has unique advantage which includes oral administration and long half-life (26 h). At higher doses of drug,sitamaquine affects parasite motility, morphology and growth.Clinical trials reported that cure rate was between the range of 87%-100% when dose was selected between 1.5 mg/kg/day and 3.0 mg/kg/day up to 28 days[74]. However, further studies are required to assess its safety issues and further development. Summary of small phaseⅠandⅡclinical trial conducted using sitamaquine is listed in Table 6.
4. Need of a new chemotherapeutic agent
As mentioned in the previous section, lots of serious loopholes and drawbacks are associated with current available chemotherapeutic regimens for VL. Therefore, there is an urgent need to develop some novel and efficacious chemotherapeutic agents against VL. Development of new chemical entity is a laborious, timeconsuming process and is also expensive. Screening of available chemotherapeutic moieties against VL is a good alternative chemotherapeutic option for VL.
4.1. Dox as a novel approach
Dox is water-soluble anthracycline topoisomerase inhibitor antibiotic. Dox is obtained from the immediate antecedent daunorubicin. Dox is a 14-hydroxylated derivative of daunorubicin which is a natural product, obtained from different Streptomyces strains, hence abundantly found in nature. Dox formulations are available as Doxil®(PEGylated liposomal type of nanoformulation),Myocet®(non-PEGylated liposomal formulation), and Caelyx. All these types of Dox are available as Ⅳ formulations[73,78-80].
Dox has unquestioned massive values against a variety of neoplastic conditions such as acute lymphoblastic leukemia, acute myeloblastic leukemia, Wilms tumor (rare kidney tumor), neuroblastoma, soft tissue and bone, breast, ovarian, transitional cell bladder, thyroid,gastric carcinoma, Hodgkin's disease, malignant lymphoma and bronchogenic carcinoma in which the small cell histologic type is the most responsive compared to other cell types. It's far frequently utilized in combination chemotherapy and as an ingredient in numerous chemotherapy regimens. Dox inhibits biosynthesis of macromolecules synthesis via DNA intercalation[81-83] and untimely blocks the topoisomeraseⅡ(which removes super coils in DNA transcription process). Therefore, it is regarded as topoisomeraseⅡinhibitor or topoisomeraseⅡpoisons.
Dox exhibits linear pharmacokinetics with wide distribution in plasma, liver, lungs, heart, and kidneys. Dox is absorbed by cells and binds to cellular components, particularly nucleic acids.Researchers reported counter intuitively activity of Dox and found that as effective as gold standard drug AmB against Leishmania parasite[84,85]. To overcome these limitations associated with the current therapeutic regimen choice, several drug delivery strategies have been accomplished by research teams and several opportunities would open a new door to explore the Dox against VL therapy.
4.2. Mechanism of Dox action against Leishmania parasite
The mammalian host redox defense framework mainly depends on glutathione oxidized and glutathione reductase and it is supplanted in Leishmania with the aid of an analogous, but unique framework depends on oxidized trypanothione [N1-N8-bis (glutathionyl)spermidine, T(SH)2] and trypanothione reductase[86,87]. The di-thiol trypanothione is a chief regulatory molecule for the DNA precursors synthesis, homeostasis of ascorbate, detoxification of hydrogren peroxides, and the sequestration/export of thiol-conjugates[88].Synthesis and reduction of trypanothione are fundamental for the upkeep of diminishing intracellular milieu, which renders the respective enzymes as attractive molecular drug targets.Trypanothione-dependent enzymes, for example, tryparedoxin peroxidase diminishes peroxides utilising electrons donated either directly from trypanothione or via the redox intermediate tryparedoxin. Trypanothione dependent hydrogen peroxide metabolism is especially significant in leishmaniasis for the reason that they lack peroxidase. Since this thiol is absent in humans and is essential for the continued existence of the parasites, these enzymes make molecular targets for the development of new drugs to treat these diseases[89]. Dox exhibits its anti-leishmanial activity by acting as competitive inhibitor for trypanothione reductase enzyme(trypanothione reductase is a redox-balancing enzyme of Leishmania spp). This enzyme is responsible for decreasing cellular oxidative stress in parasite. Dox competes for trypanothione reductase enzyme by acting as subversive substrates and inhibits enzymatic action.Anti-leishmanial potential of Dox is being exploited using numerousdrug delivery strategies. Macrophage target oriented delivery of Dox is an ideal choice of drug delivery strategies for effective management and treatment of VL. Targeted delivery to intra-cellular regions of macrophages by encapsulation of Dox into numerous carriers and biopolymers provides a more efficient way to treat VL.Several attempts of Dox delivery against VL have been performed and summarized in Table 7 and exclusively discussed in the abovementioned section.
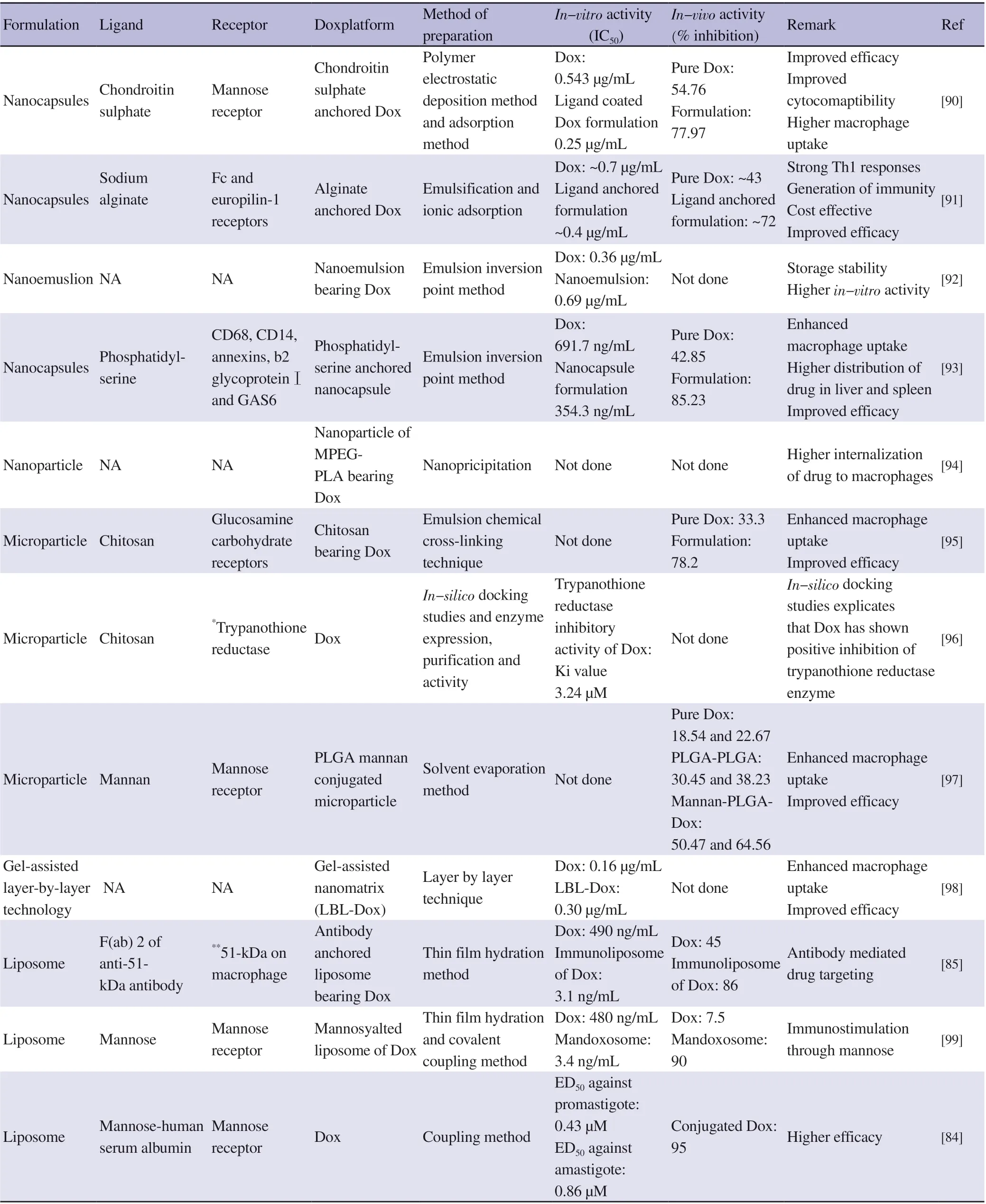
Table 7. Summary of various doxorubicin formulations use for visceral leishmaniasis treatment.
4.3. Chemotherapy of VL by Dox
Optimum chemotherapy and management of VL have not attended even after availability of a variety of chemotherapeutic agents due to emergence of resistance, nephrotoxicity and cardiotoxicity. Thus,alternative option is required due to continuation in dose-dependent negotiation with patient health as well as emergence of resistance and treatment failure. There is an emerging alarm for innovation of a new alternate and vital option for chemotherapy of VL. Chemotherapy of VL by Dox is an outcome of ongoing research for identification of possible alternates of current available chemotherapeutic agents against VL.
Dox shows inhibitory potential against trypanothione reductase,which is typically presented in Leishmania spp. compared to human.In 1992, Sett et al. was the first research group presented in-vitro and in-vivo therapeutic efficacy results of Dox against amastigotes and promastigotes forms of parasite. Results described the Dox efficacy through an in-vitro activity (IC50was 0.43 μM and 0.86 μM against amastigotes and promestigotes, respectively) and in-vivo activity in infected mouse model at dose of 625 μg/kg/day up to four consecutive days showing 95% spleen parasite burden reduction[84].In another study, novel gel nanomatrix of Dox showed improved efficacy against L. donovani. In this case, IC50of developed Dox formulation (0.16 μg/mL) was 1.9 folds lesser than that of plain Dox(0.30 μg/mL), while cell viability study demonstrated that unloaded nano-matrix was safer against macrophages represented by more than 90% cells viability[84,100].
Trypanothione reductase is a key enzyme and a principal thiol in Leishmania parasite and analogue of glutathione reductase, which is present in mammals. Trypanothione reductase accounts for removal of reactive oxygen species (ROS) by detoxifying the hydroperoxides,converting them into alcohol and water through tryparedoxin protein.Active sites of glutathione reductase and trypanothione reductase are different, hence they exhibit quiet specificity towards their substrates.Therefore, trypanothione reductase is considered to be a selective marker for anti-parasitic drugs development. Shukla and co-workers have explored the trypanothione reductase inhibitor ability of Dox.Authors acquired computational (in silico docking studies) and experimental approaches (MTT assay, hemolysis, in-vitro activity)to explore Dox efficacy against leishmaniasis[101]. Molecular docking studies revealed that tetracyclic (anthracyclin) moiety of Dox acts through hydrophobic interaction and is responsible for interaction with amino acids of the hydrophobic patch (Try21,Met113 and Glu467) of the active site. Dox showed binding energy comparable to potent inhibitors. Therefore, these compounds may be a potential inhibitor of trypanothione reductase. At concentration of 25 μM, it showed approximately 60% decline in total thiol in comparison to control group. Intracellular ROS measurements using CM-H2DCFDA probe indicated that ROS levels were sufficiently enhanced when promastigotes were incubated with 20 μM Dox as estimated by an increase in fluorescence intensity. Increment in fluorescence intensity with respect to enhanced concentration of Dox represented the increased levels of ROS leading to oxidative stress.In-vitro efficacy assessment of Dox demonstrated the promising results in term of IC50against promastigotes (11.76±0.11) μM and axenic amastigotes (12.02±0.15) μM, respectively. Kansal and co-workers developed a nanoemulsion core loaded nanocapsules entrapping Dox and efficacy was evaluated against L. donovani. Dox loaded nanocapsules were efficiently internalized into macrophages indicating enhanced efficacy. IC50of developed Dox nanocapsules was almost 1.9 folds compared to plain drug against intracellular amastigotes[102].
4.4. Chemo-immunotherapy of VL in association with Dox
Absence of parasite specific cell-mediated immune response is a distinctive immunological aspect of active VL infection[103].The equilibrium of specific T cell subsets, cytokines, macrophage activation, and the induction of nitric oxide (NO) sets up the impact of illness. It has been accounted that during infection, there is a raise in the number of IL-4-positive neutrophils, NK cells, and IL-10 monocytes, while the number of IFN-g-positive, IL-2, and IL-12 eosinophils is significantly diminished. But serum levels of TNF-α and IL-6 were high in patients with active VL[104,105]. Conversely,cured cases have shown a typical type 1 response with an increment in IFN-γ, IL-2 neutrophils, eosinophils, NK cells and with an increase in IL-12 monocytes. Helper and cytotoxic T cells are known to play a vital role in the immune response to this infection,connecting the innate immune response to the development of efficient adaptive cellular immunity mainly through IL-2 and IFN-γ production. These two cytokines drive the effect or functions of macrophages and elicit a Th1 immune response. These findings recommend that any intervention which can shift the immune response from Th2 type to Th1 type will have a major role in cure and prevention of VL. Lots of scientific approaches have been performed to develop an immunotherapeutic formulation of Dox for successful management of VL.
Active targeting of Dox is achieved by encapsulating it in mannosefabricated liposomes and synergistic activities of INF-γ were studied in a BALB/c mice model by Kole et al[106]. L. donovani infected macrophages were treated with free drug Dox, doxosome(liposomal Dox), and mandoxosome (mannose coated liposomal Dox). Representative IC50values of manodoxosome, free Dox and doxosome were 3.4, 480 and 9.6 ng/mL, respectively. Prior INF-γ treatment showed synergistic activity represented by 55%, 1% and 44% suppression of infection in macrophages by manodoxosome(1 ng/mL), doxosome (3 ng/mL) and free Dox (150 ng/mL),respectively. In L. donovani infected mice model, treatment with various forms of Dox resulting in 50% reduction of infection when combined with INF-γ. These results remarked the synergistic inhibitory effect of INF-γ with Dox for suppression of parasitic burden[103,106].
Kansal and co-workers studied the immunotherapeutic potential of sodium alginate coated nanocapsules loaded with Dox. They developed sodium alginate anchored nanocapsules bearing Dox for an immunomodulatory assisted anti-leishmanial activity. Developed formulation showed 1.5 folds higher cellular internalization followed by significantly enhanced in-vitro and in-vivo efficacy in J774A cells and infected hamster.
Sodium alginate is a potent substance having an excellent property to activate macrophages and also an inducer of releasing cytokines and cytotoxic agents[91]. Alginate coated nanocapsules of of Dox uptaken by J774A.1 macrophages was improved by 1.5 folds when compared with uncoated nanocapsules. Alginate coated nanocapsules of Dox showed higher efficacy than uncoated Dox nanocapsules against intra-macrophagic amastigotes. Coated formulation treatment exhibits enhanced apoptotic efficiency as compared with nanoemulsion of Dox and free Dox as evidenced by cell cycle flow cytometric analysis, and decline in mitochondrial membrane potential, ROS and NO production. In-vivo results demonstrated that the increased levels of iNOS, TNF-α, IFN-γ and IL-12 when treatment was given with alginate coated nanocapsules of Dox compared with Dox nanoemulsion. Parasitic burden was diminished in Leishmania infected hamsters treated with alginate coated formulation in comparison to uncoated formulation.
Mukherjee et al. designed and fabricated an actively targeted immunotherapeutic formulation of Dox (immune-doxosome) using 51 kDa proteins (a parasite specific protein isolated from infected macrophages)[85]. Immuno-doxosomes were prepared by grafting F(ab)2 of anti-51 kDa antibody onto the liposomal surface bearing Dox via covalent bonding interaction mechanism. Time-dependent cellular internalization was studied with infected and normal macrophages cells, which revealed that phagocytosis of immunedoxosomes by infected macrophages reached equilibrium at 30 min,whereas uptake of immune-doxosome by normal macrophages was significantly lesser and independent of time was shorter. Higher internalization of immunodoxosomes ultimately assured higher activity with lesser non-specific tissue toxicity of Dox. In-vitro activity of immune-doxosomes within macrophage was compared with free Dox, doxosomes and non-specific doxosomes. IC50of immune-doxosomes (3.1 ng/mL) was significantly lesser compared with free Dox (490.0 ng/mL), doxosomes (9.8 ng/mL) and nonspecific immune-doxosomes (8.0 ng/mL). Immunodoxosomes showed 100% suppression of parasite in spleen with subsequent decline in spleen weight to normal value when treatment was given with a dose equivalent to 250 μg/kg/day of the Dox. On the other hand, free doxorubicin showed only 45% reduction in splenic parasite burden. Doxosome and non-specific immuxodosome caused 84% and 86% reduction in infection, respectively. These results demonstrated that immunodoxosomes were more effective than doxosome, non-specific immune-doxosomes and free Dox for suppressing parasite infection in macrophages and infected animals[85].
4.5. Macrophages targeted delivery of Dox
VL parasite invades, resides and multiplies in macrophages of mammalian host cell. Mastcell presenting cell plays a fundamental role in the etiology of VL and it is a major target for the treatment of VL. The ultimate strategy and goals of formulation development for targeting mastcell presenting cell are crucial. It could usher an unorthodox but highly effective therapeutic paradigm for treatment of VL with envisioned targeting of antigen presenting cells (macrophages) to overcome the limitation of current available marketed options. Macrophages express diverse ranges of engulfment receptors that are able to bind modified polysaccharides,lipoproteins, senescent and apoptotic cells, and a range of polyanionic molecules. Several approaches have been already tried on the basis of receptor ligand delivery of Dox to achieve a macrophage targeted delivery limiting non-specific tissue toxicity. Sett and coworkers developed and utilized mannosylated neo-glycoprotein carrier for facilitating receptor-mediated endocytosis of Dox[84].Here, mannose-human serum albumin was conjugated with Dox.Result of in-vitro activity suggested that conjugated Dox was found to be more efficiently eliminated (12.5 times) from intracellular amastigotes of L. donovani in peritoneal macrophages compared to free drugs. Furthermore, conjugated Dox greatly eliminated splenic intracellular parasites burden when treatment was given in four consecutive dosages (5 μg/kg/day) up to 45 days[99].
Similarly, mannose conjugated polylactic-co-glycolic acid microparticles loaded with Dox was prepared and evaluated against experimental VL for selective macrophages targeting of liver and spleen[97]. In another study, chitosan anchored micro-particulate system with a vision for vectoring Dox to reticuloendothelial system via passive targeting through particle size and active targeting by receptor ligand association concept. In-vivo activity in syrian golden hamsters showed that ligand anchored microparticles have significant higher reduction (78.2±10.4)% in parasite load compared to free Dox (33.3±2.4)%[97].The diblock biodegradable co-polymer bearing Dox nanoparticles were prepared using nano-precipitation method for macrophage targeting. Developed nanoparticles were evaluated for macrophage targeting potential and obtained results depicted the higher internalization of nanoparticles in macrophages as compared to free drug. Slow release of Dox from nanoparticles resulted in availability of Dox for longer time at the parasitic site of infection, thus leading to greater efficacy[95].
Etiology of macrophage based clearance of apoptotic cells is reconciled by phosphatidylserine that is present on the apoptotic cell external surface and gets exposed which executes binding through macrophages scavenger receptors, CD-68 receptor, CD14, annexin,β-2 glycoproteinⅠand GAS6. Macrophage targeting potential of phosphatidylserine was explored and evaluated. Authors developed a phosphatidylserine anchored nanocapsules dosage form using layer-by-layer method for efficient macrophage targeting of Dox.Developed cargos were evaluated during in-vitro and in-vivo conditions and subsequently compared with uncoated nanocapsules and free Dox. Cellular internalization of phosphatidylserine anchored nanocapsules in J774A.1 macrophage was 1.75-fold higher than plain nanocapsules. In-vitro activity of phosphatidylserine anchored nanocapsules having Dox was performed against L. donovani intramacrophagic amastigote and results were expressed as inhibition of parasite growth (IC50) observed after 48 h of incubation. The respective IC50values of phosphatidylserine anchored nanocapsules,uncoated nanocapsules and free Dox were (354.33±5.13),(391.67±20.40) and (691.67±10.41) ng/mL, respectively. Results of in-vivo antileishmanial activity showed that phosphatidylserine anchored nanocapsules caused (85.23±4.49)% inhibition of the splenic parasite burden, while uncoated nanocapsules and free Dox caused (72.88±3.87)% and (42.85±2.11)% parasitic inhibition respectively in L. donovani infected hamsters[94]. Chaurasia et al.developed chitosan anchored nanoparticles bearing AmB and Dox to specifically target macrophage with two different antileismanial agents. Cellular uptake study in macrophage cell line revealed that chitosan anchored formulation internalized 2.02 fold higher than uncoated formulation. Developed combined drug delivery anchored with chitosan showed significantly (P<0.05) lower IC50against L. donovani infected macrophages in comparison to uncoated formulation[93].
4.6. Drug repurposing of non-PEGylated liposome encapsulated doxorubicin for VL
4.6.1. Composition of non-PEGylated liposome encapsulated doxorubicin
Non-PEGylated liposome encapsulated doxorubicin consists of single unilamellar liposomes of size approximately 160 nm which is composed of egg phosphotidyl choline: cholesterol (in the molar ratio 55:45). It is commercially available as a package of 3 vials.First vial contains doxorubicin HCl-50 mg and lactose NF-250 mg; second vial contains empty liposomes in citrate buffer, e.g.phosphotidyl choline-142.6 mg and cholesterol NF-57.4 mg in citrate buffer (57.6 mg/mL)-2 mL; and the third vial buffer for injection contains sodium carbonate anhydrous 54.6 mg in 3.1 mL of water for injection. After preparation as mentioned in the patient information leaflet[93-95], the doxorubicin is almost completely encapsulated into the empty liposomes through active loading process, i.e., through pH gradient established across the liposomal membrane (acidic pH ~4.5 inside the liposomes) to produce 2 mg/mL liposomal doxorubicin solution. The drug to lipid ratio is approximately 0.25:1 (wt:wt). The resulting solution is red-orange and opaque in appearance and the final pH of the formulation is 6.5 to 8.5.
4.6.2. Passive targeting of reticuloendothelial systems by non-PEGylated liposome encapsulated doxorubicin
It was concluded that in comparison to Doxil®(PEGylated liposomal formulation of doxorubicin) and doxorubicin plain solution, the non-PEGylated liposomal version of doxorubicin(Myocet®) is rapidly or actively taken up by the cells of the reticuloendothelial systems like macrophages and monocytes and ends up in large quantities in liver and spleen compared to other tissues. This phenomenon can be capitalized to passively target doxorubicin to intracellular parasites (VL) of macrophages in liver and spleen. The opsonisation of non-PEGylated liposomes drives phagocytosis by macrophages and drives their passive targeting into these reticuloendothelial systems. It is also interesting to note the concentration of doxorubicin in the heart tissue for both liposomal formulations (Doxil®and Myocet®) was lower compared to free doxorubicin solution which directly reflects on the lower incidence of cardiotoxicity of the liposome encapsulated doxorubicin.
Non-PEGylated liposome encapsulated doxorubicin can passively target macrophage resident parasites in VL with reduced cardiotoxicity. Hence, authors strongly propose re-purposing of non-PEGylated liposome encapsulated doxorubicin for treating VL as an alternative to other liposomal formulations like liposomal AmB.
5. Conclusions
Pharmaceutical research on various anti-leishmanial chemotherapeutic agents and their variable responses in diverse geographical areas generated an urge to explore the novel therapeutic strategy to alleviate severe and diverse VL. However, priority should be given to remedies which are safer, more inexpensive and with minimal toxicity than existing prescription medicines. Substantial progress in search for Dox as a novel chemotherapeutic agent mainly depends upon the initiative of individual academic laboratories to consortia and public-private partnerships. Various biopolymers and carriers also potentiate the anti-leishmanial activity of Dox via more efficient apoptosis and would provoke the host body's immune response.Combination of immune stimulation with anti-leishmanial drug is more effective than treatment alone with a chemotherapeutic. Non-PEGylated liposome encapsulated doxorubicin, which has potential to passively target macrophages and its intracellular parasite, can be re-purposed to treat VL. Hence, this potential therapeutic approach may encourage future validation or repurposing of the other drugs for treatment of VL infection.
6. Future outlook
Leishmaniasis is a great challenge and a global burden in terms of drug discovery and drug delivery due to capability of parasite residing in cells and subsequently widespread to other areas in the body. Higher toxicity, emergence of MDR and variable responses challenged the scientists to develop new anti-leishmanial drugs as well as new strategies to control it. The future perspectives of VL treatment depend on the development of optimum anti-leishmanial Dox formulation alone or with various biopolymers which could control the vectors, monitor the efficiency of given therapeutic regimen and diagnosis method with more sensitivity and specificity.Complete eradication of leishmaniasisis also depends on combined chemotherapy of doxorubicin based drug delivery systems with other drugs, so that addition and synergism property of drugs will curb the emergent MDR effectively.
Conflict of interest statementNo conflict of interest was reported by the authors of this article.There is no conflict of interest between the authors and manufacturer.
Acknowledgments
Authors would like to acknowledge Dr. Pankaj K. Singh,Department of Pharmaceutics, National Institute of Pharmaceutical Education & Research (NIPER), Hyderabad for his guidance in writing this manuscript.
Funding
The authors received no extramural funding for the study.
Authors’contributionsS.R.S. developed the theoretical formalism and final version of the manuscript. R.N.S supervised the work.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Hurdles in achieving the goal of malaria elimination by India
- Prevalence and factors associated with belief in COVID-19 vaccine efficacy in Indonesia: A cross-sectional study
- Mosquito larva distribution and natural Wolbachia infection in campus areas of Nakhon Ratchasima, Thailand
- Genetic variation of sand flies (Diptera: Psychodidae) in Gampaha and Kurunegala districts of Sri Lanka: Complementing the morphological identification
- A rare presentation of Guillain-Barre syndrome with GQ1b positivity: A case report