极小种群馨香玉兰特异性单核苷酸多态性位点开发*
2022-08-23张涛李学张竞印轩鹏贺水莲
张涛,李学,张竞,印轩鹏,贺水莲
(云南农业大学 园林园艺学院,云南 昆明 650201)
极小种群野生植物(plant species with extremely small populations,PSESP)指分布地域面积狭小或成间断分布,且长期以来遭受各类不同外界自然环境因素的胁迫影响,已经远远小于稳定生存范围界限内的最小生存种群,且随时都有可能发生濒临灭绝的野生植物种类[1]。馨香玉兰(Magnoliaodoratissima)为木兰科(Magnoliaceae Juss.)木兰属(MagnoliaLinn.)常绿乔木,其花洁白芳香,枝繁叶茂,有较强的净化环境和抗污染能力,为优质的观赏树木[2]。馨香玉兰仅产于云南省,主要分布于文山壮族苗族自治州的广南、麻栗坡和西畴等地,其所有居群都分布在自然保护区外,缺乏针对性的保护。馨香玉兰的野生资源仅剩下5个居群,均处于高大乔木之下,光照不足,生长前景很不乐观,结实率低[3];种子中存在抑制发芽的物质,且抑制效果显著[4]。由此可见其种群生存状况堪忧。其濒危级别已属极危,并列入我国Ⅱ级重点保护野生植物[5],也是我国云南特有的极小种群物种,急需开展保护。而今,已有学者对其种群生态学[6-8]、保护生物学[9]、林学[10]、育苗[11-12]等方面开展了研究。然而,对于馨香玉兰的分子标记开发、保护遗传学研究还很薄弱。随着分子生物学的发展,开发分子标记来对濒危植物进行遗传评价,从而提出精确保护策略,是保护濒危植物的有效手段。
SLAF-seq(specific-locus amplified fragment sequencing)技术是通过构建SLAF-seq数据库,将具有特异性长度的片段(SLAF标签)筛选出来并进行高通量测序,最后将得到的符合质量要求的SLAF片段作为目标物种的全基因组信息,进而依据这些特异性SLAF标签能够在全基因范围内迅速鉴定准确性高的SNP变异位点信息,利用特异性位点可以构建遗传连锁图谱、系统进化树、PCA分析、基因流分析等用于遗传进化和群体结构分析。SLAF测序技术在保护遗传学领域已经得到了广泛利用,并取得了一定的成果。如:Zheng等[15]基于SLAF-seq技术对红橘×枳(Citrusreticulata×Poncirustrifoliata)杂交群体进行了遗传多样性分析、Xia等[16]基于SLAF-seq技术对油棕(Elaeisguineensis)进行了遗传多样性和群体结构的评价、Chang等[17]基于SLAF-seq技术对侧柏(Platycladusorientalis)进行了遗传保护研究。此类工作证实了SLAF-seq测序技术可以提供大量的SNPs以供群体遗传学分析[18]。本研究以5个野生居群和2个栽培居群共70份馨香玉兰资源为材料,采用SLAF-seq建立文库,通过筛选特异性相关位点扩增片段,在全基因组范围之内开发大量稳定性高、特异性强的基因SNP位点,可为馨香玉兰的保护遗传学、谱系地理学及种群生态学等研究提供分子标记。
1 材料与方法
1.1 实验材料
选取云南省文山州西畴、麻栗坡及广南的5个野生居群以及云南省昆明市金殿公园、昆明植物园的2个栽培居群共70份馨香玉兰资源为实验材料(表1)。采集其无病斑的新鲜叶片,用硅胶干燥。每个居群采集10个个体,个体间距离不低于10 m。
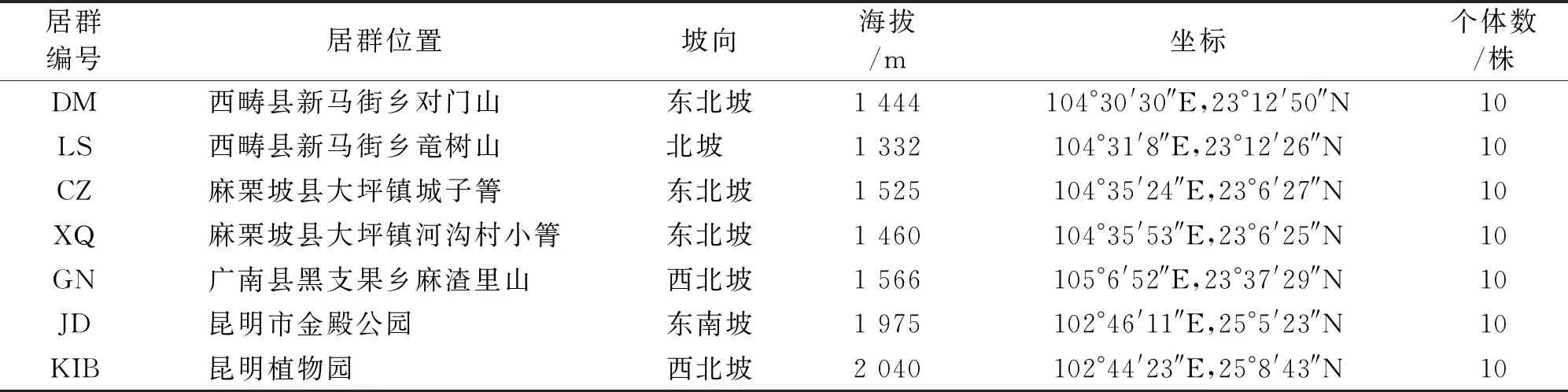
表1 7个馨香玉兰居群的地理位置和取样情况
1.2 DNA的提取
采用改进的CTAB法[21]提取馨香玉兰样品的基因组DNA,用NanoDrop 1000分光光度计评估所得DNA的浓度和质量,并使用2%SDS-PAGE显示结果;然后将DNA样品稀释至100 ng/μL,用于随后的SLAF-seq技术分析。
1.3 酶切建库
由于馨香玉兰的基因组数据尚未有研究,因此无法确定其基因组结构大小及GC浓度等相关信息。本研究对70个馨香玉兰个体分别进行建库处理。首先选取近缘种鹅掌楸(Liriodendronchinense)基因组(ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/003/013/855/GCA_003013855.1_ASM301385v1/GCA_003013855.1_ASM301385v1_genomic.fna.gz)作为参考基因组进行酶切预测(鹅掌楸基因组大小为1.56 Gb、GC含量为39.06%),然后利用RsaI+EcoRV-HF©限制性内切酶组合进行酶切,对获得的酶切片段(SLAF标签)进行3′端加A处理、连接Dual-index[22]测序接头、PCR扩增、纯化、混样、切胶选取目的片段,经文库质检合格后用Illumina平台完成检测。
1.4 Illumina HiSeqTM2500测序及产出数据的质量分析
经文库质检合格后用Illumina HiSeqTM2500进行测序。测序后获得的序列通过去接头、去低质量阅读框和去污处理方法,最后获得到干净序列。为充分保证评估后本次酶切实验结果的正确性,采用粳稻(Oryzasativassp.japonica)作为酶切实验结果对照,进行了相同的数据处理参与建库和测序。为有效保证每个样本序列测序的数据质量,对测序后所得的样品序列数据进行GC比例和Q30数据分析。
1.5 SLAF标签的获得和SNP 标记的开发
利用BWA软件[23]将序列测序reads比对在不同参考基因组上,统计不同序列染色体上的SLAF标签和多态性SLAF标签。按照不同序列之间的相识程度,对不同馨香玉兰样本的序列进行比对聚类,最后聚集在一起的reads来源于同一个SLAF标签,不同SLAF标签间的相似度相对于同一SLAF标签在不同样品间的序列相似度较低。一个SLAF标签的不同样品间序列有差异(即有多态性),被称之为多态性SLAF标签。通常使用GATK[24]和SAMTOOLS[25]两种筛选方法可以组合开发出SNP,将两种筛选方法结合得出的SNP标记交集作为最终正确的SNP标记数据集,筛选的标准值为MAF>0.05。
2 结果与分析
2.1 建库评估
电子酶切预测以鹅掌楸为参考基因组,采用组合限制性内切酶RsaI+EcoRV-HF©进行酶切,将酶切片段为414~464 bp的序列定义为SLAF标签,共得到127 393个SLAF酶切标签(表2)。通过对粳稻检测数据的评估监控,从而判断酶切方法的有效性。粳稻基因组大小为374.30 Mb,并通过SOAP[26]软件将粳稻的测序reads与其参考基因组进行比对,比对后的结果见表3。经比对,双端比对效率在90.39%(一条序列两端在参考基因组上的比对跨度介于500~1 000 bp的reads占总数的比例),比对效率基本正常。样品测序碱基分布情况见图1。

表2 酶切预测确定的酶切方案信息统计

表3 粳稻测序reads比对结果统计
图1 馨香玉兰测序碱基分布
酶切效率是简化基因组测序是否成功的关键性指标。基因载体上的结构区域(如环状结构域、连续酶切位点等)复杂、基因组DNA样品纯度较低、酶切综合利用持续时间短和质量水平不足等这些客观因素均有可能直接地影响限制性内切酶的活力,从而造成酶切位点识别不准确而未能完全切开。根据reads插入片段中剩余酶切位点的具体检测结果比例进行测算,其比例越高,酶切效率越好[27]。粳稻数据的酶切效率统计结果见表4。粳稻对照组的酶切效率为91.46%,表明酶切反应正常。90.39%的双端比对效率和91.46%的酶切效率说明SLAF建库正常。

表4 粳稻数据酶切效率评估统计
通过对照双端比对序列在基因组中的定位,可以估算SLAF标签的实际长度,并绘制对照读长插入片段的长度分布图(图2),从而估计实际的片段选择范围。序列的插入片段长度都在预期的选择范围之内(图2),说明序列测序数据质量正常,采用的这种测序分析方法可信度高[28]。
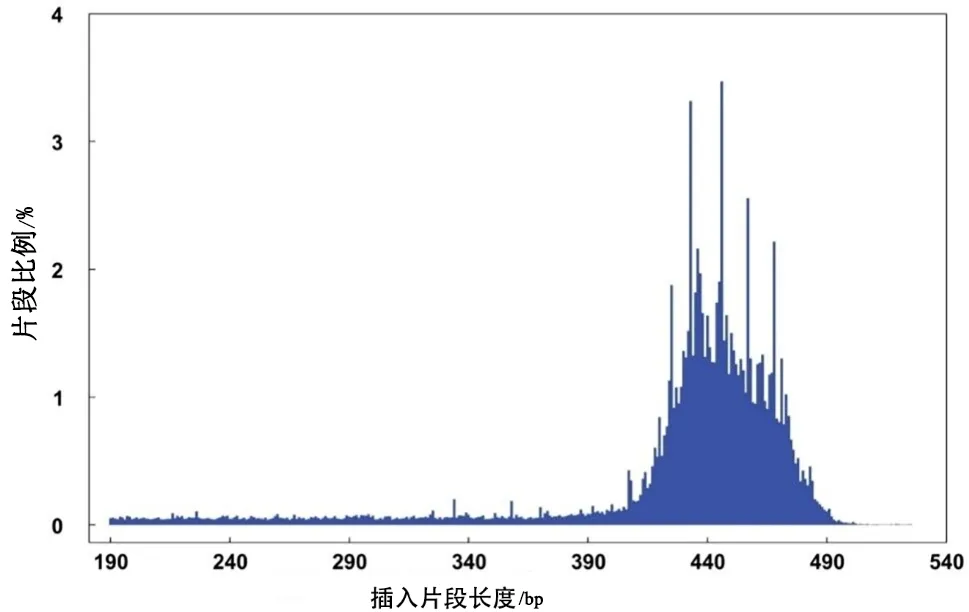
图2 对照序列插入片段分布
2.2 测序数据统计与评估
为提高综合分析的结果质量,以读长100 bp×2 的数据作为后续的实验数据进行综合评估,以粳稻的数据为参考评估馨香玉兰资料库的数据准确性。通过Illumina HiSeqTM2500平台测序,总共得到了229.54 Mb reads的数据(表5),在各个实验样本中所统计得到的数据读长长度值的范围为1 499 485~7 274 846个。其中:来自昆明金殿的JD4样本所获得数据量最大,为7 274 846个读长;西畴县对门山的DM7样本数据量最小,为1 499 485个读长。样本测序所获得的GC比例为41.5%~44.38%,其中GC比例的最大值44.38%为来自广南县的GN7样本、GC比例的最小值41.5%为来自西畴县竜树山的LS7样本、GC比例的平均值为43.01%。由此可见,GC比例普遍较低,说明已达到测序条件。测序质量值 Q30 的范围为78.54%~93.22%,平均值为86.96%。其中:Q30测序的最小值来自于麻栗坡县小箐的XQ2样本,其值仅为78.54%;来自于昆明植物园的KIB9样本的Q30测序值最大,为93.22%,每个样本的Q30值都在70%以上。表明测序技术的碱基错误率非常低,所获数值均合格。
2.3 SLAF 标签与 SNP 标记的开发
通过序列分析,从70份馨香玉兰样本的基因组中共获得843 082个SLAF标签。标签的平均测序深度约是11.78 ×(表5),其中多态性SLAF的标签有287 843个。对获得的多态性SLAF标签进行分析,共得到2 998 167个馨香玉兰群体SNP标记,各样品中SNP标记数量为476 062~1 392 468个,各样品检测到的SNP完整度为15.87%~46.44%、杂合率为3.42%~10.69%。筛选去除完整度≤50%以及次要基因频率≤5%的SNP,总共获得了180 650个高度一致性的群体SNP位点,占SNP总量的6.03%。
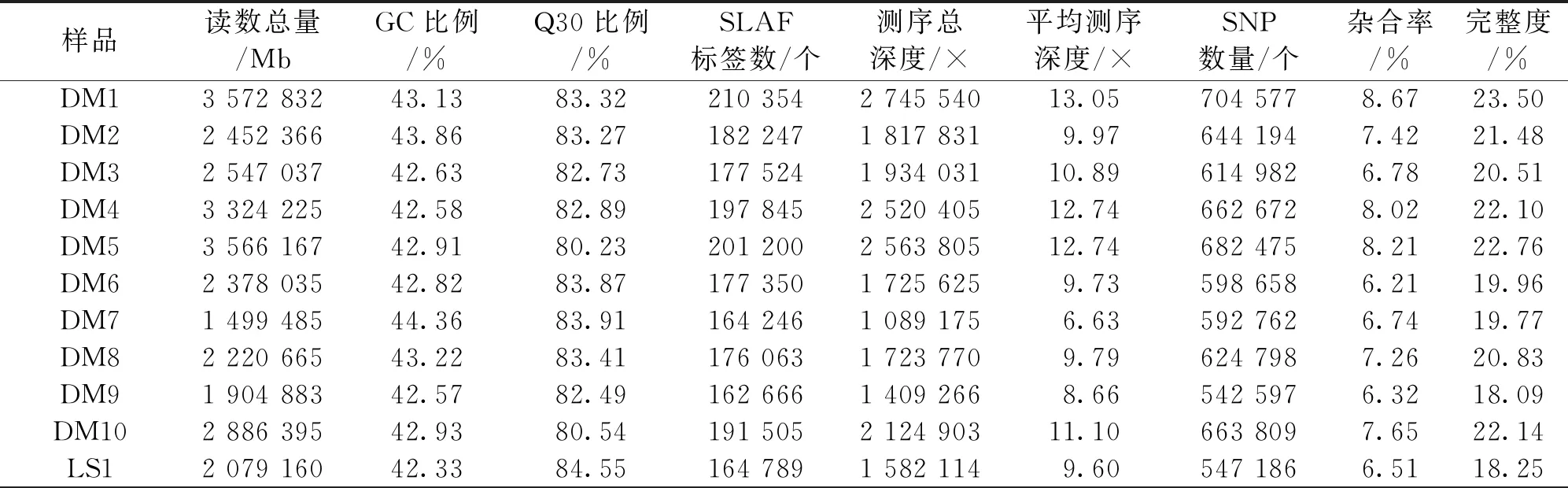
表5 馨香玉兰SLAF标签统计和SNP 信息统计及对照测序结果
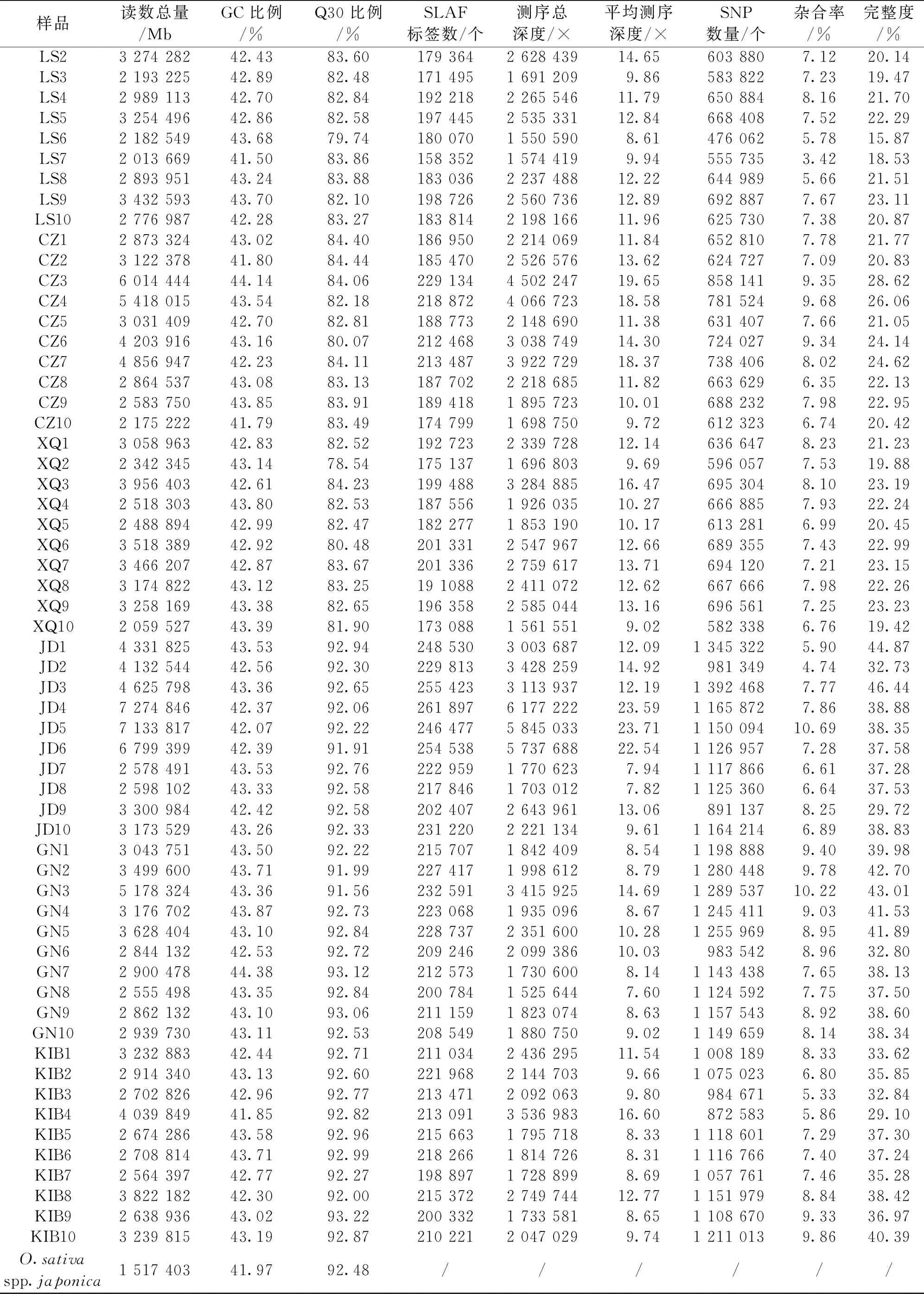
续表5
2.4 遗传多样性分析
基于180 650个高质量SNP位点,对馨香玉兰5个野生居群及2个栽培居群进行遗传多样性检测(表6)。结果显示:7个馨香玉兰群体的观测杂合度(Ho)范围在0.136 3~0.355 1之间,其中最高的是XQ群体,最低的是JD群体;期望杂合度(He)的范围在0.296 1~0.357 4之间,其中最高的是KIB群体,最低的是GN群体;Shannon指数(I)的范围在0.455 3~0.535 9之间,其中最大值是KIB群体为0.535 9,最小值是GN群体为0.455 3;Nei多样性指数的范围在0.312 6~0.386 3之间,其中最大值是KIB群体为0.386 3,最小值为GN群体为0.312 6。7个馨香玉兰群体在群体水平上都具有较为丰富的遗传多样性,其中KIB群体的遗传多样性水平相对较高,GN群体的遗传多样性水平相对较低。KIB为栽培居群,其遗传多样性指数相对处于较高水平,推测其个体可能来源于不同的野生居群。
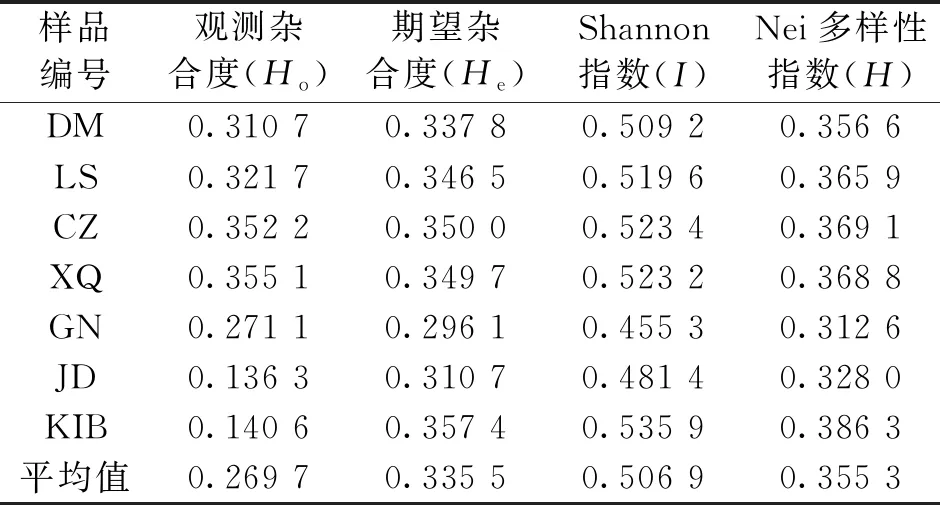
表6 馨香玉兰的遗传多样性指数
3 讨论与结论
目前,我国在极小种群野生植物保育方面已做了大量工作,云南省在极小种群野生植物保育方面的成绩尤为突出。随着记录了云南101种极小种群野生植物的主要识别特征、分布现状、受威胁因素和主要保护建议等信息的图书——《云南省极小种群野生植物保护名录(2021版)》[29]的出版发行,将更进一步地促进云南极小种群野生植物的保育工作。随着测序技术的发展,传统的分子标记已无法满足研究需求,越来越多的研究者开始采用高通量测序技术来进行极小种群的保护遗传学研究。马永鹏等[30]为弄清漾濞槭(Aceryangbiense)极小种群的形成与维持机制,更好地指导保护工作,对漾濞槭105个个体开展了重测序,从遗传分化、基因流、遗传多样性、种群历史、有害突变和近交等方面对漾濞槭进行了深入研究。杨丰懋等[31]对显脉木兰(Magnoliafistulosa)进行dd-RAD简化基因组测序,对显脉木兰的遗传多样性,种群历史及第四纪冰期的有效居群大小等进行了研究,对显脉木兰提出了精确保护策略。由此可见,使用高通量测序深层次挖掘物种的遗传多样性、种群结构及历史、有害突变等信息具有明显的优势。
馨香玉兰基因组庞大而复杂,目前尚无全基因组序列信息公开,开发传统分子标记耗时耗力,不能满足馨香玉兰保护遗传学研究需求。因此,本实验运用SLAF-seq技术对该物种的特异性分子标记进行开发,并对其遗传多样性参数进行分析,结果显示这些SNP标记在馨香玉兰不同群体中表现出较为丰富的多态性,与金蕊等[9]利用RAPD技术对馨香玉兰进行遗传多样性测定的结果(Shannon指数为0.568 6、Nei’s指数为0.390 3)相比,本研究得到的遗传多样性指数稍低,因为RAPD技术的稳定性较差,是显性遗传,不能识别杂合子位点,这使得遗传分析相对复杂,在遗传多样性计算时会因显性遮盖作用而使计算位点间遗传距离的准确性下降,而使用SLAF-seq,稳定性和准确性较高。
馨香玉兰被列为第一批极小种群物种,但跟其它木兰科植物相比,如大果木莲(M.grandis)[32](I=0.365 1,H=0.243 3)、乐昌含笑(Micheliachapensis)[33](I=0.475 1,H=0.325 5)等,其遗传多样性水平却相对较高(I=0.5069,H=0.355 3)。这说明馨香玉兰虽然片段化明显,仅剩的种群小,但仍具有较强的生存力,导致其濒危的原因可能是外界环境的因素,如生存环境恶劣,上层乔木抢夺生存资源[33],而不是由物种本身遗传多样性单一导致。本研究探索利用SLAF-seq开发分子标记,既可为馨香玉兰未来的种群结构、遗传进化与种群演化历史等提供高质量的分子标记,同时也可为馨香玉兰分子辅助选育、高密度遗传连锁图谱的建立等方面提供参考。
