硫酸卡那霉素及注射液的有关物质测定方法研究
2022-08-19刘照振寇晋萍刘海涛侯金凤齐麟黄晓春李彧刘琦车宝泉北京市药品检验研究院国家药品监督管理局仿制药研究与评价重点实验室中药成分分析与生物评价北京市重点实验室北京102206
刘照振,寇晋萍,刘海涛,侯金凤,齐麟,黄晓春,李彧,刘琦,车宝泉(北京市药品检验研究院 国家药品监督管理局仿制药研究与评价重点实验室 中药成分分析与生物评价北京市重点实验室,北京 102206)
氨基糖苷类抗生素主要对金黄色葡萄球菌和需氧革兰阴性杆菌,包括铜绿假单胞菌有强大的抗菌作用;对沙雷菌属、布鲁杆菌、沙门杆菌、嗜血杆菌、痢疾杆菌以及结核分枝杆菌、其他分枝杆菌属亦具有良好的抗菌作用。氨基糖苷类抗生素优点较多,如水溶性好、抗菌能力强、性质稳定、吸收排泄较好等,因此在临床上应用较为广泛[1]。卡那霉素(kanamycin)是1958年日本明治制果药业株式会社开发生产的由α-去氧链胺衍生物组成的氨基糖苷类抗菌药,对革兰阴性菌及青霉素、链霉素、红霉素等产生耐药性的金黄色葡萄球菌、大肠埃希菌、产气杆菌、肺炎杆菌和痢疾杆菌有很强的抗菌作用[2]。目前各国药典对卡那霉素均有收录[3-7],其原料药根据硫酸盐数目不同分为单硫酸卡那霉素(C18H36N4O11·H2SO4)和硫酸卡那霉素(C18H36N4O11·xH2SO4)两种[8]。据文献报道,卡那霉素主成分为卡那霉素A,发酵和纯化工艺中会产生卡那霉素B、卡那霉素D等副产物和脱氧链霉胺降解产物[9-10]。
卡那霉素缺乏特征的紫外吸收,因此一般采用蒸发光检测器(HPLC-ELSD)法对其进行质量控制。《中国药典》2020年版[7](Ch.P2020)收载的硫酸卡那霉素原料及硫酸卡那霉素注射液有关物质测定方法均为HPLC-ELSD法,仅对杂质卡那霉素B进行检查,未对其他杂质进行控制,并且也有文献[10]采用ELSD方法对其他杂质的控制进行研究,由于ELSD要求流动相具有挥发性,使得色谱系统的优化受到限制,因此分离能力较差。除《中国药典》2020年版外,各国药典均采用薄层色谱法进行有关物质检查,方法专属性较差且分离能力较低,无法对杂质谱进行深入分析。近几年已有文献[10-14]相继报道,采用高效液相色谱-柱后衍生化-荧光检测法对硫酸卡那霉素滴眼液、硫酸异帕米星注射液、妥布霉素滴眼液等氨基糖苷类制剂的质量控制方法展开研究。因此,针对硫酸卡那霉素原料及注射液需要开发新的质量控制方法。本方法重点参考文献[10-14],在原有文献报道的高效液相色谱-柱后衍生化-荧光检测法的基础上,调整色谱系统和柱后衍生化系统,增加系统载样量,提高检测灵敏度和杂质分离度,新验证的方法相比《中国药典》2020年版和文献方法,杂质的分离更好,检测灵敏度和测定准确度进一步提高,可更好地控制硫酸卡那霉素原料及注射液的质量。
1 试验材料
1.1 试验仪器 LC-20A高效液相色谱仪(岛津公司);RF-20A荧光检测器(岛津公司);waterse2695高效液相色谱仪(美国沃特斯公司);2000ES蒸发光散射检测器(ELSD)(奥泰公司);Pickering PCX 5200柱后衍生化系统(美国沃特斯公司);XA205电子天平(Mettler-toleto公司)等。
1.2 药品与试剂 硫酸卡那霉素对照品(批号:130556-201502,中国食品药品检定研究院);卡那霉素B对照品(批号:130548-200501,中国食品药品检定研究院);硫酸卡那霉素样品A(A公司,批号:180601、180602、181103),单硫酸卡那霉素样品B(B公司,批号:20120112-1、20120203-1),单硫酸卡那霉素样品C(C公司,批号:D1405020、D1405022),硫酸卡那霉素注射液样品D(D公司,批号:1306171、1306172),硫酸卡那霉素注射液样品F(F公司,批号:13091301、13091303);氢氧化钠、硼酸、冰醋酸、2-巯基乙醇(分析纯)、无水硫酸钠由国药集团化学试剂有限公司提供(分析级);邻苯二甲醛(色谱纯)由北京百灵威科技有限公司提供;三氟醋酸由J&K CHEMICL提供(色谱级);三氟乙酸、乙腈、甲醇均为色谱纯,购自Merck公司;庚烷磺酸钠(色谱纯)由北京百灵威科技有限公司提供;水为高纯水。
2 方法与结果
2.1 色谱条件 色谱柱:Agilent ZORBAX SB C18柱(4.6mm×250mm,5μm);流动相:pH3.4缓冲液(取庚烷磺酸钠一水合物8.7g和无水硫酸钠32g,加水溶解并稀释至2000mL,用冰醋酸调节pH值至3.4±0.1)-甲醇(95∶5)为流动相A,以甲醇为流动相B,按表1进行线性梯度洗脱;柱温:35℃;流速:1.0mL/min;荧光检测器:激发波长340nm,发射波长455nm;进样体积:5μL。配有柱后衍生化装置,衍生化试液:取邻苯二醛0.8g、甲醇300mL、2-巯基乙醇2ml和硼酸缓冲液(取72.0g硼酸和43.0g氢氧化钠,加水溶解并稀释至4000mL,用1mol/L硼酸溶液或1mol/L氢氧化钠溶液调节pH值至10.4±0.1)700mL,混合,摇匀,置棕色瓶中,避光储存;衍生化温度:35℃;衍生化试液流速:0.5mL/min。

表1 流动相梯度洗脱条件
2.2 溶液的制备 有关物质测定溶液:精密称取硫酸卡那霉素适量或精密量取硫酸卡那霉素注射液适量,加水溶解并定量稀释制成约含卡那霉素1mg/ml的溶液,作为有关物质供试品溶液。取卡那霉素对照品适量,精密称定,加水溶解并定量稀释成约含卡那霉素2.5、5、10、15和20μg/mL的溶液,作为对照溶液(1)、(2)、(3)、(4)和(5),用线性回归方程以自身对照法计算有关物质含量。
2.3 系统适用性溶液 系统适用性试验溶液:取卡那霉素B对照品约10mg,置100ml量瓶中,加水稀释至刻度,摇匀,作为卡那霉素B对照品贮备液;另取卡那霉素对照品B约10mg,置10mL量瓶中,精密量取卡那霉素B对照品贮备液1ml,置同一量瓶中,振摇使其溶解,加水稀释至刻度线,摇匀,作为系统适用性溶液。
将系统适用性溶液照“2.1”项下条件分析,与采用《中国药典》2020年版中的HPLC-ELSD法比较,结果详见图1与图2,如图2所示采用新的方法,卡那霉素峰与卡那霉素B,及各杂质峰之间分离度更能满足药典要求。
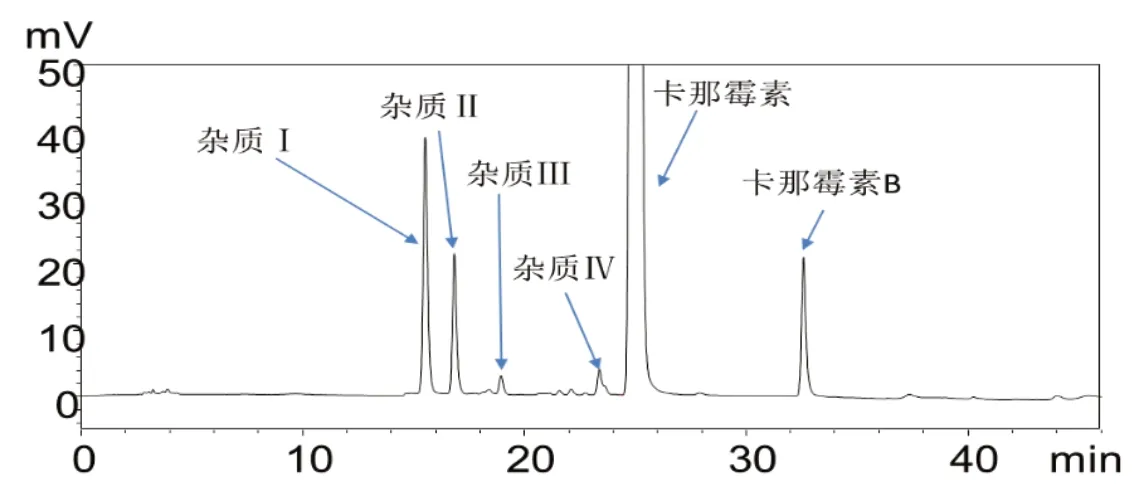
图1 HPLC-荧光检测法系统适用性典型色谱图
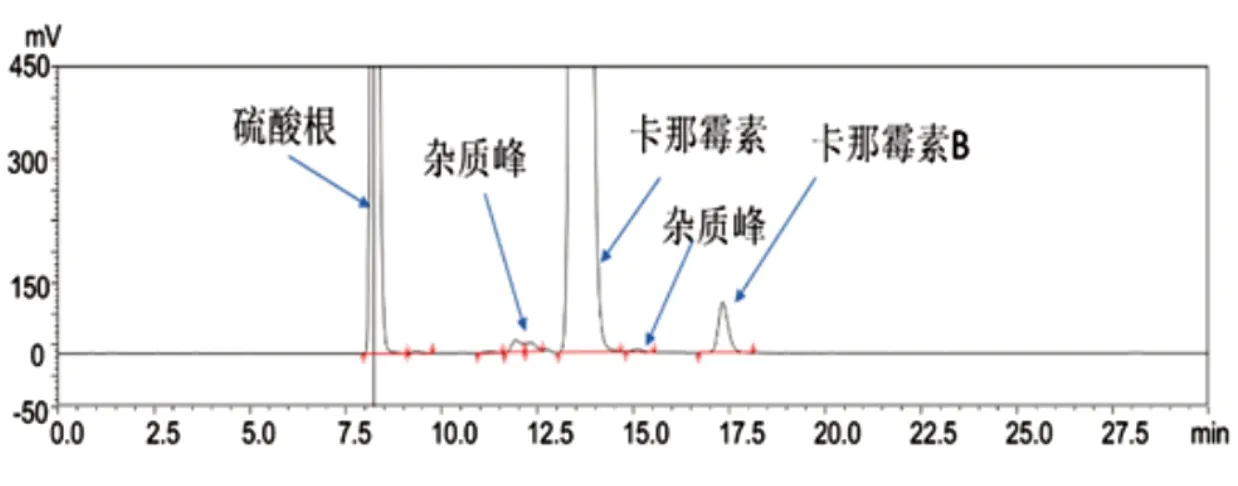
图2 《中国药典》2020年版HPLC-ELSD法测定系统适用性典型色谱图
2.4 专属性试验 按企业提供的制剂处方,称取亚硫酸氢钠约2.0g、依地酸二钠0.1g,置同一1000ml量瓶中,加入适量纯化水使溶解,另加入浓硫酸27.6ml,加水稀释至刻度,摇匀,精密量取1ml,置250ml量瓶中,加水稀释至刻度摇匀,作为辅料干扰溶液。将辅料干扰溶液和空白溶剂照“2.1”项下条件分析,结果详见图3。如图所示,空白与辅料均不干扰样品测定,方法专属性良好。
图3 专属性试验色谱图。注:A.空白;B.辅料
2.5 破坏试验 取D企业批号为1306171的样品,精密量取5份,各1mL,分别置250mL量瓶中,进行如下处理:①酸破坏:加2mol/mL盐酸溶液5mL,60℃水浴加热12h,加2mol/mL氢氧化钠溶液5mL中和;②碱破坏:加2mol/mL氢氧化钠溶液5mL,60℃水浴加热12h,加5mol/mL盐酸溶液1mL中和;③光破坏:在5000lx照度下放置12h;④热破坏:60℃水浴加热12h;⑤氧化破坏:加6%过氧化氢溶液5mL,60℃水浴加热12h。将上述5种破坏的溶液用水稀释至刻度,摇匀,按“2.1”项下条件进样分析,色谱图详见图4。结果表明,在硫酸卡那霉素光照和加热破坏下均稳定,在碱和氧化条件下,降解杂质Ⅱ和杂质Ⅲ的含量略有上升,在酸性破坏中降解杂质Ⅱ和杂质Ⅲ的含量有明显上升。由结果可以看出,卡那霉素在各条件破坏下得到的杂质均能实现满意分离。
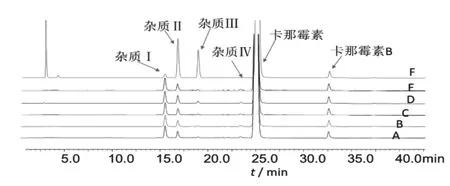
图4 样品原液及5种方式破坏样品溶液的色谱图。注:A.未破坏;B.加热破坏;C.光照破坏;D.氧化破坏;E.碱破坏;F.酸破坏
2.6 线性关系考察 精密称定硫酸卡那霉素对照品适量,用水溶解并稀释制成约1.0mg/mL的卡那霉素溶液,作为线性储备液。分别精密量取线性储备液适量,制成系列浓度的线性溶液,作为线性考察用溶液。分别取线性考察溶液5μL,注入色谱仪,记录色谱图。以卡那霉素峰面积y对质量浓度(x)进行线性回归,得出回归方程:y=43.415x-15.627(r=0.9998)结果表明卡那霉素在0.005-1.0mg/mL浓度范围内,与卡那霉素峰面积呈良好线性。
2.7 检测限和定量限 荧光方法的最低检测浓度为0.3μg/mL(LOD,S/N=3),最低定量浓度0.5μg/mL(LOQ,S/N=10),按进样量5μL换算,得到方法的检测限为1.5ng,定量限为2.5ng,分别相当于供试品浓度的0.03%和0.05%。
2.8 重复性考察 取批号为1306171的样品,按2.2项下方法配制供试品溶液各6份,照2.1项下的方法色谱条件,在10h内每2h进样,分别测定其已知杂质、其他单个最大杂质和总杂质的量,结果详见表2:卡那霉素B的RSD(n=6)为0.5%,杂质Ⅰ的RSD(n=6)为0.4%,杂质Ⅱ的RSD(n=6)为0.8%,杂质Ⅲ的RSD(n=6)为1.3%,杂质Ⅳ的RSD(n=6)为1.0%,总杂质的RSD(n=6)为0.3%,表明重复性良好。
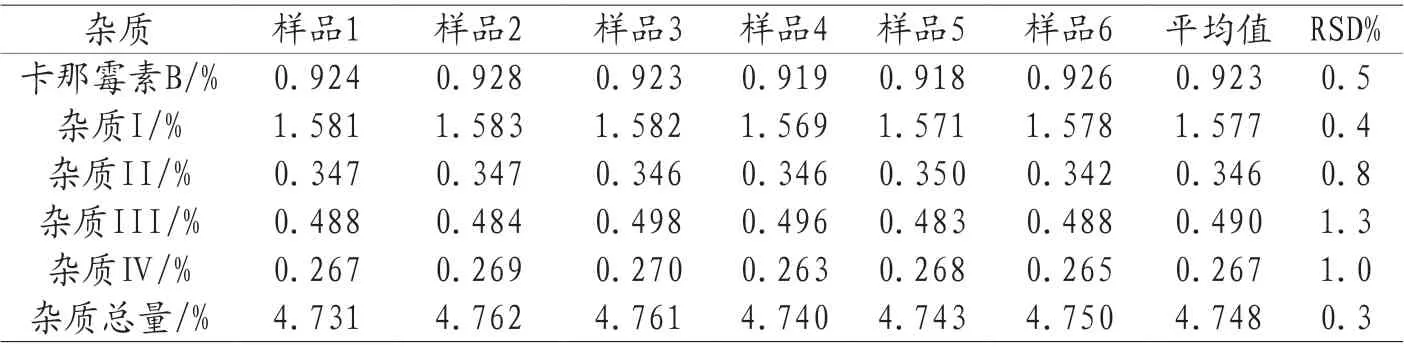
表2 重复性试验结果
2.9 耐用性试验 对方法的耐用性进行分析,分别改变流速、柱温、流动相pH值和相同色谱柱的批次,以卡那霉素峰与卡那霉素B峰之间、卡那霉素与杂质Ⅳ、杂质Ⅱ分别与杂质Ⅰ、杂质Ⅲ之间分离度衡量分离情况,耐用性试验结果见表3。由结果可见,最优条件为1.0mL/min流速,35℃柱温,流动相为pH值为3.4。
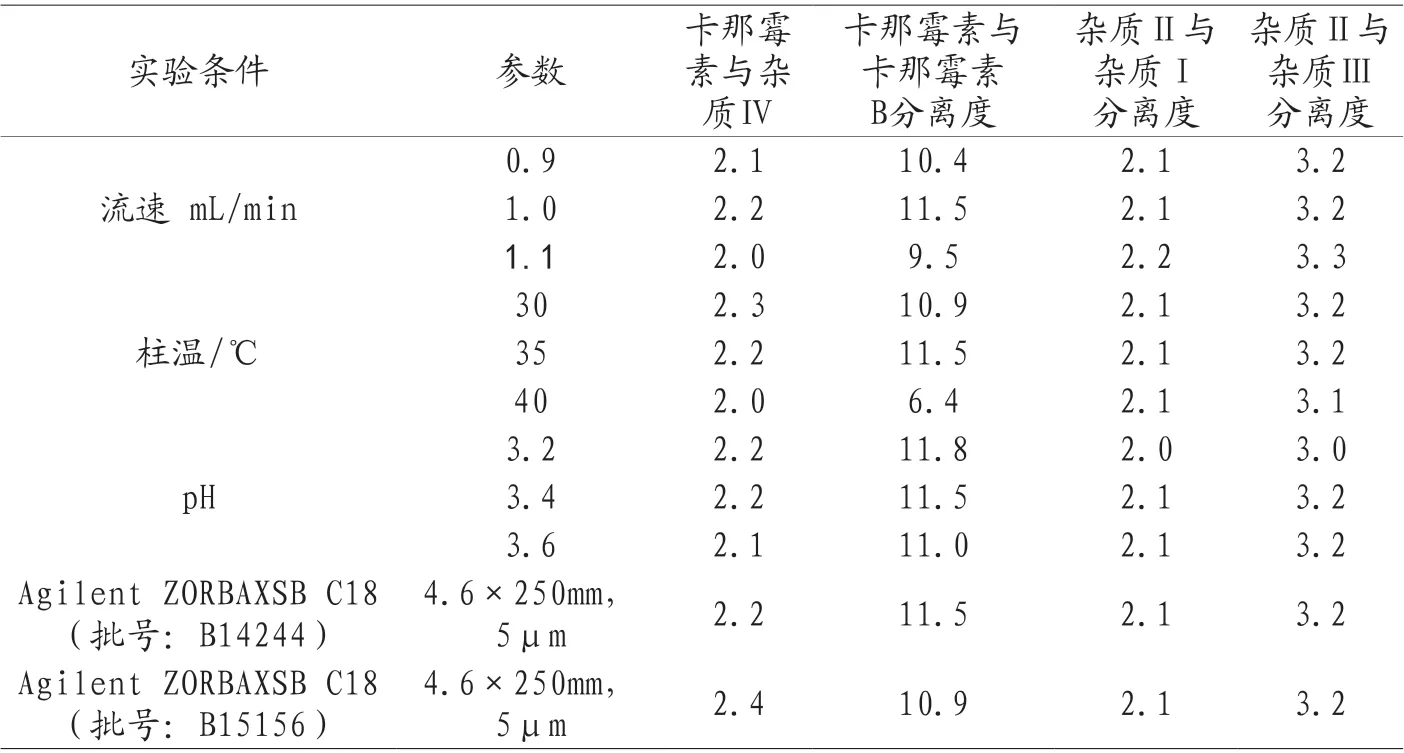
表3 耐用性试验结果
2.10 有关物质检查结果 分别取各企业硫酸卡那霉素原料及注射液,按“2.2”项下方法配制有关物质供试品溶液及有关物质对照品溶液,按照“2.1”项下条件分析。计算杂质含量,结果见表4。
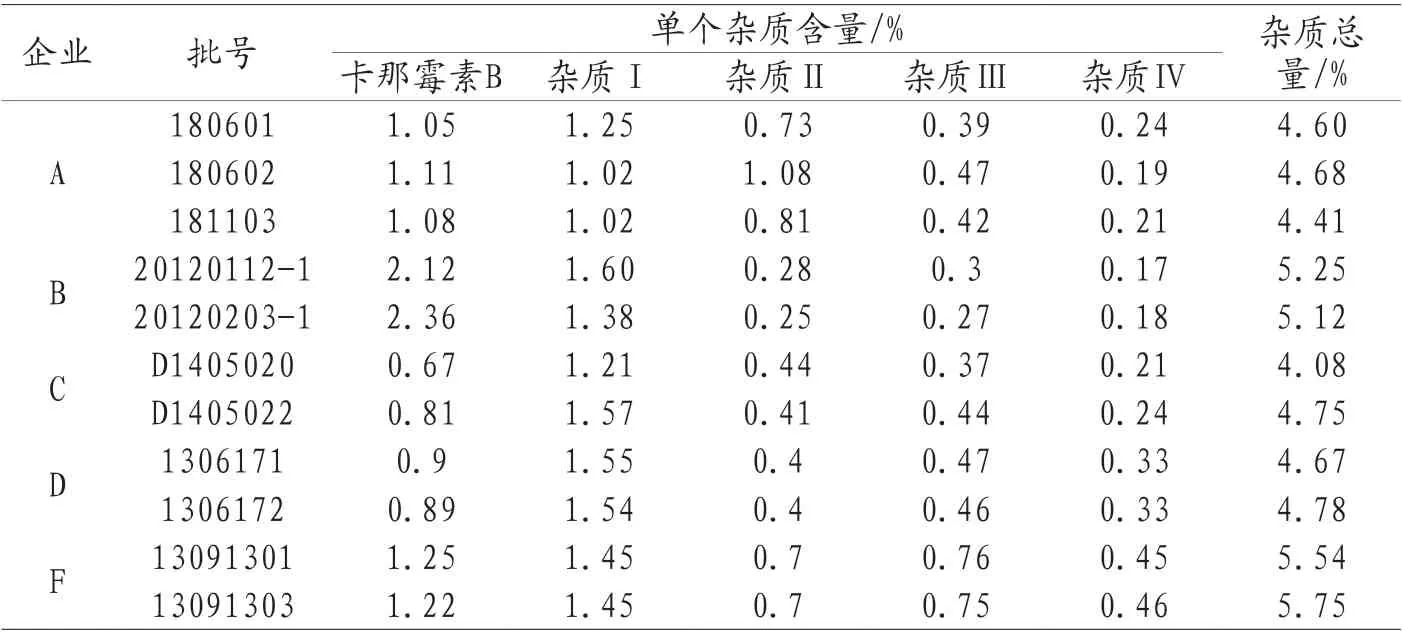
表4 有关物质测定结果
3 讨论
3.1 色谱条件选择 本文重点参考硫酸卡那霉素滴眼液质量控制相关文献[12-14],采用高效液相色谱-柱后衍生化-荧光检测器法,新建硫酸卡那霉素原料及注射液有关物质的测定方法。在已有文献方法的基础上,调整色谱系统和衍生化条件。
在液相色谱条件方面:在不改变流动相中缓冲盐的情况下,进一步调整流动相中缓冲盐和乙腈的比例,并相应地调整梯度洗脱时间程序,色谱柱由(4.6mm×100mm,3.5μm)的短柱,改变为Agilent ZORBAX SB C18(4.6mm×250mm,5μm)长柱。
柱后衍生化条件:在不改变柱后衍生化试剂、缓冲盐等条件下,充分考察了衍生剂的流速、衍生化的温度、衍生化试剂的用量和衍生剂的pH值等方面的因素。在衍生剂的流速方面,衍生剂流速影响衍生化率,在流速0.3mL/min、0.4mL/min、0.5mL/min时主峰面积无明显变化,当衍生剂流速增大到0.6-1.0mL/min时,主峰面积呈现减小趋势,但是各杂质分离度随衍生剂流速增大而提高,因此综合考虑各色谱峰分离度和衍生化率的因素,衍生剂流速确定为0.5mL/min;衍生化试剂邻苯二甲醛用量方面,衍生剂加入量在1.2g/L、1.0g/L、0.9g/L、0.8g/L和0.7g/L时各峰面积无明显变化,当衍生剂加入量减少至0.6g/L和0.5g/L时,各峰面积呈现减小趋势,因此综合考虑衍生化率,衍生剂加入量确定为0.8g/L;考察了衍生剂不同pH值时(10.2、10.4、10.6)和衍生化温度(30℃、35℃、40℃)各峰面积的变化,温度和pH值的变化对峰面积几乎无影响,故选择与文献相同的衍生化温度为35℃和pH值10.4。
3.2 杂质定位及计算方法选择 中检院提供的卡那霉素B为定性鉴别用对照品,缺乏纯度值,杂质Ⅰ、杂质Ⅱ、杂质Ⅲ和杂质Ⅳ各国药典均未有相关对照品,无法进行杂质回收率的准确度试验,也无法按杂质对照外标法或带校正因子的自身对照法计算。此外,《中国药典》2020年版也采用不带校正因子的自身对照法,其他各国药典均采用自身对照的薄层色谱法进行有关物质检查,因此受各国药典无对照品的限制,新建方法也采用不带校正因子的自身对照法计算。
3.3 新建方法与《中国药典》现行标准有关物质测定结果的比较 用新建硫酸卡那霉素及注射液有关物质的测定方法与《中国药典》2020年版收载硫酸卡那霉素有关物质测定方法HPLCELSD,分别对企业提供样品进行测试,测试样品的代表性图谱详见图5和图6,样品测定结果详见表5。
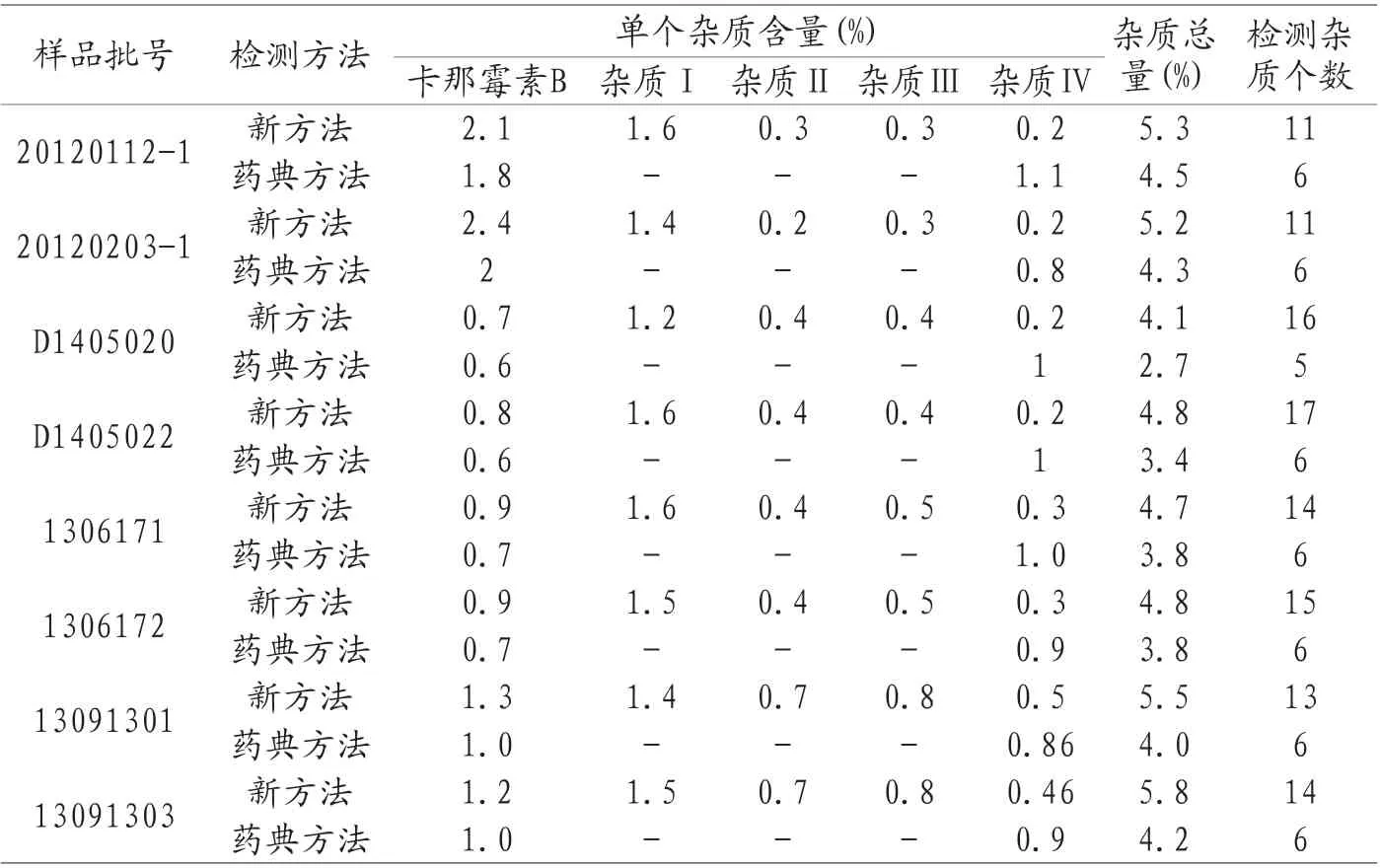
表5 HPLC-柱后衍生化-荧光检测法(新拟定方法)和HPLCELSD(中国药典方法)有关物质测定结果对比
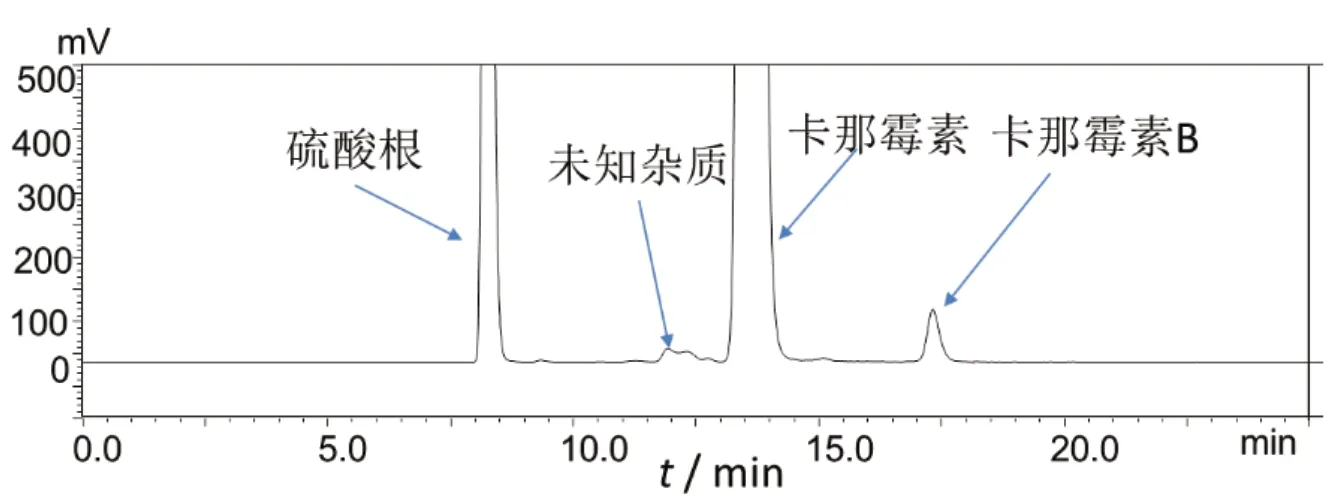
图5 HPLC-ELSD系统有关物质测定法供试品溶液实验色谱图
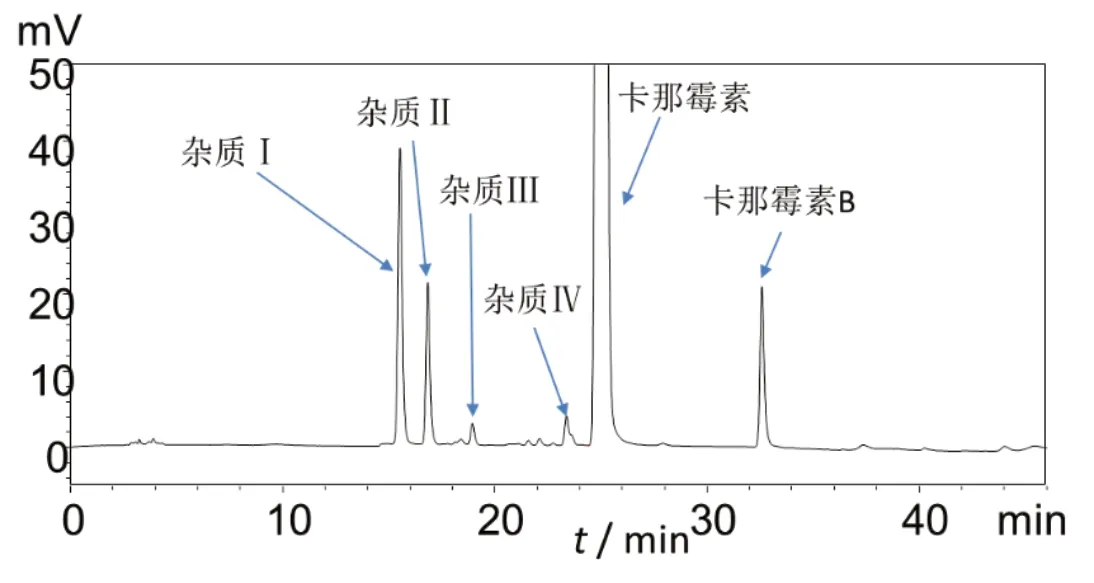
图6 HPLC-柱后衍生化-荧光检测系统有关物质测定法供试品溶液实验色谱图
根据测定结果,HPLC-FLD法检出杂质数量更多(见表5);对于样品中主要的降解杂质Ⅱ与杂质Ⅲ和工艺杂质Ⅰ,HPLC-柱后衍生化-荧光检测法都能实现更好的分离和检测(见图6),因此,本法更能真实、客观、有效地反映出硫酸卡那霉素的质量。
4 小结
本研究是国家药典委员会关于硫酸卡那霉素原料及硫酸卡那霉素注射液药品标准提高项目,本文所建立的HPLC-柱后衍生化-荧光检测法较HPLC-ELSD法(《中国药典》2020年版)提高了分离度和灵敏度,新方法测定硫酸卡那霉素原料及注射液的有关物质,检出杂质数量更多,结果更加准确。因此,本文建立的HPLC-柱后衍生化-荧光检测法更能有效控制硫酸卡那霉素原料及注射液的质量。
