高效分子排阻法测定注射用头孢唑肟钠聚合物的方法学研究
2022-08-18孔令洋孙巧巧李正国
孔令洋 韩 勇 孙巧巧 王 冉 李正国▲
1.山东省济宁市食品药品检验检测研究院,山东济宁 272027;2.中国中医科学院中药研究所,北京 100700
头孢唑肟钠属于第三代头孢菌素,用来治疗各种敏感菌导致的疾病,如尿路感染、呼吸道感染、腹腔感染等,疗效较好,具广谱抗菌作用[1-2]。作为头孢菌素的一种,其聚合物的形成可能会引起各种不良反应,如过敏、休克等,而聚合物杂质在药品的生产、贮存、运输、使用任一过程中均可能产生[3-6]。本品的执行标准中聚合物的测定采用葡聚糖凝胶G10色谱法[7],该方法具有分析时间长,灵敏度低,不能使用有机溶剂,主峰与聚合物峰不能有效分离等缺点,难以准确反映药品的质量情况。近几年,高效分子排阻色谱法广泛应用于分析抗生素中的聚合物[8-14]。此方法准确度、灵敏度高,简便易行。本研究使用TSK gel G2000SWXL亲水硅胶色谱柱,建立的高效分子排阻法测定注射用头孢唑肟钠的聚合物的方法,分析时间短,可大幅提高检测效率,操作方便,检测结果更为准确。
1 仪器和试药
安捷伦1200系列高效液相色谱仪(配备紫外检测器),岛津LC20A液相色谱仪(双波长紫外检测器);赛默飞TSQ Quantum Access MAX液质联用仪 东曹TSK gel G200SWXL色谱柱(300 mm×7.8 mm,1.80 μm),葡聚糖凝胶G10(40~120 μm)色谱柱(内径1.0~1.4 cm,柱长30~40 cm)头孢唑肟对照品(批号130504-201503,中国食品药品检定研究院,纯度97.2%);乙酸铵(分析纯),磷酸氢二钠(分析纯),磷酸二氢钠(分析纯),甲醇为色谱纯,均购于国药集团化学试剂有限公司。
2 方法与结果
2.1 色谱条件
用0.075 mol/L pH 7.0的磷盐缓冲液[0.075 mol/L磷酸氢二钠溶液-0.075 mol/L磷酸二氢钠溶液(61∶39)]作为流动相A;水为流动相B,流速为 1.5 ml/min,检测波长为254 nm,拟建立高效分子排阻色谱法色谱条件:0.075 mol/L磷酸盐缓冲液[0.075 mol/L磷酸二氢钠溶液-0.075 mol/L磷酸氢二钠溶液(39∶61)]-乙腈(90∶10),检测波长为254 nm。
2.2 对照品溶液的制备
精密称取头孢唑肟对照品约10 mg,置100 ml容量瓶中,加水溶解并稀释至刻度,摇匀,作为对照品溶液。
2.3 供试品溶液的制备
精密称定样品(相当于头孢唑肟0.5 g),置100 ml容量瓶中,加水溶解并稀释至刻度,摇匀,作为供试品溶液。
2.4 流动相的选择
分别以0.075 mol/L磷酸盐缓冲液[0.075 mol/L磷酸二氢钠溶液-0.075 mol/L磷酸氢二钠溶液(39∶61)]-乙 腈(95∶5)、0.075 mol/L磷 酸 盐缓冲液[0.075 mol/L 磷酸二氢钠溶液-0.075 mol/L 磷酸氢二钠溶液(39∶61)]-乙腈(90∶10)和0.075 mol/L 磷酸盐缓冲液[0.075 mol/L 磷酸氢二钠溶液-0.075 mol/L 磷酸二氢钠溶液(61∶39)]-甲醇(95∶5)为流动相进行分离效果比较。结果流动相为0.075 mol/L磷酸盐缓冲液[0.075 mol/L磷酸二氢钠溶液-0.075 mol/L磷酸氢二钠溶液(39∶61)]-乙腈(90∶10)时,在主峰前可分离出2个杂质峰,且分离效果最好。
2.5 专属性实验
2.5.1 系统适用性试验 取供试品溶液10 ml,加入0.1 mol/L 氢氧化钠溶液1 ml,放置于室温,10 min后,再加1 ml的0.1 mol/L 盐酸溶液中和,充分摇匀,取100 μl,注入液相色谱仪,记录色谱图;头孢唑肟峰与其前相邻降解杂质峰间的分离度要符合实验要求。
2.5.2 加速破坏实验 酸破坏:取供试品溶液10 ml,加0.1 mol/L盐酸1 ml,室温放置1 h,取20 μl注入液相色谱仪,记录色谱图。
碱破坏:取供试品溶液10 ml,加0.1 mol/L氢氧化钠溶液1 ml,室温放置1 h,取20 μl注入液相色谱仪,记录色谱图。
热破坏:取供试品溶液10 ml,在60℃水浴中加热1 h,放冷,取20 μl注入液相色谱仪,记录色谱图。
254 nm紫外破坏:取供试品溶液10 ml,在254 nm的紫外光放置1 h,取20 μl注入液相色谱仪,记录色谱图。见图1。
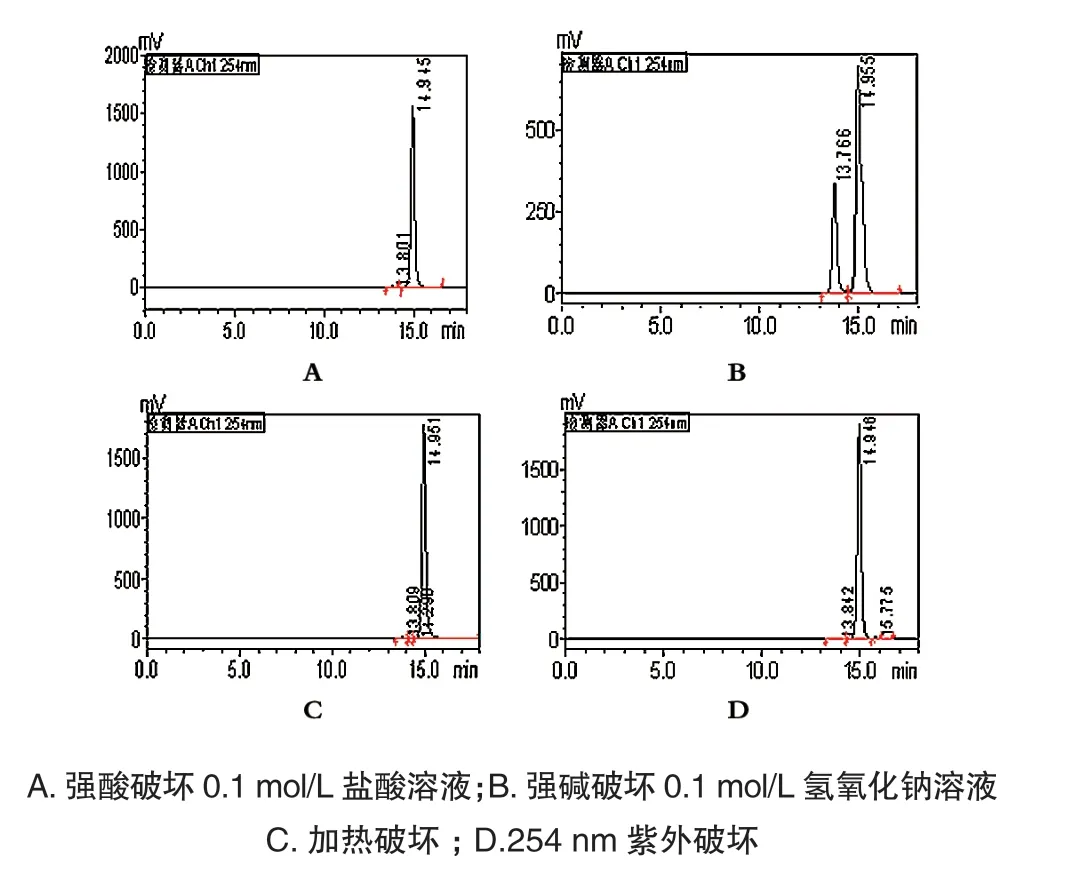
图1 HPLC色谱图
专属性试验结果显示,供试品在经过4种条件的激烈破坏处理后,其中聚合物含量均有增加,增加含量由低到高依次为经紫外破坏、酸破坏、高温破坏、碱破坏,经四种破坏处理后的聚合物峰均能完全分离,表明在该色谱条件下,头孢唑肟的降解产物均能与主峰有效分离,方法专属性良好。
2.6 线性考察
取头孢唑肟对照品12.95 mg,置于500 ml容量瓶中,加水溶解,稀释至刻度,摇匀,作为对照品储备液。精密量取适量对照品溶液1、2,5、10、20、25、50 μl进入液相色谱仪,以对照品的进样量(x),峰面积(y),进行线性回归,得到线性方程y=9104302.0196x-2640.1968(n=9),对照品溶液在质量范围0.02518~2.5180 μg时,线性关系良好。
取适量注射用头孢唑肟钠内容物,精密称定,按“2.2”方法制成含头孢唑肟供试品溶液,浓度分别 为0.5、1.0、2.0、5.0、10.0、20.0和50.0 mg/ml。色谱条件同“2.1”项,进行液相分析,横坐标为供试品溶液的浓度c、纵坐标为聚合物的峰面积A,绘制曲线,线性拟合,得到标准曲线方程为A=387137c+4741.3,结果表明:供试品溶液在浓度0.5~50 mg/ml时线性关系良好。
2.7 头孢唑肟钠的稳定性考察
取注射用头孢唑肟内容物2.0 g,置100 ml容量瓶中,加水溶解并稀释着刻度,摇匀。取20 μl注入液相色谱仪,记录色谱图。
取注射用头孢唑肟钠2.00 g,置500 ml容量瓶中,加0.90%氯化钠注射液溶解并稀释至刻度,摇匀。取20 μl注入液相色谱仪,记录色谱图。并考察将供试品溶液放置在避光和漫反射条件下高分子杂质的变化情况。见表1。
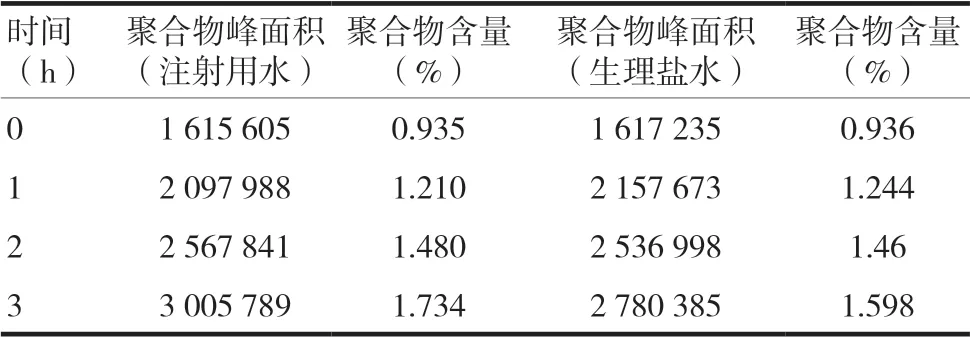
表1 头孢唑肟钠在不同介质中聚合物的稳定性考察
由药品说明书可知,本品临床上主要采用0.9%氯化钠注射液和注射用水等介质溶解样品,并要求在0.5~2 h内使用完。故本实验以按临床高剂量在上述2种介质中的考察稳定性,试验表明,注射用头孢唑肟钠溶液状态不稳定,为指导临床合理用药提供依据,供试品溶液应临用现配。
2.8 检测限和定量限
取“2.7”项下对照品溶液,逐步稀释,色谱条件按“2.1”项下,进样分析,结果显示,检测限为0.25 μg/ml[信噪比(S/N)为3.3],定量限为0.75 μg/ml(S/N=11.2),灵敏度良好。
2.9 考察供试品溶液
在不同亲水硅胶色谱柱的保留情况,发现杂质个数与保留时间有差异,经过分析,应是色谱柱内的填料对不同分子量的保留有差异。见图2~3。
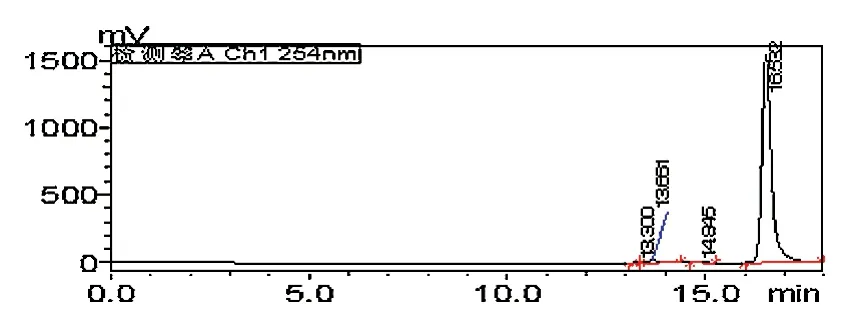
图2 供试品溶液在sepax亲水硅胶色谱柱的色谱图
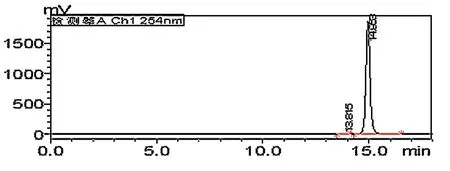
图3 供试品溶液在TSKgelG2000SWXL亲水硅胶色谱柱的色谱图
3 讨论
供试品溶液中聚合物峰与其他杂质峰在不同亲水硅胶色谱柱的保留情况,发现杂质个数与保留时间略有差异,分离机制除与分子量有关外,与离子浓度等因素相关。葡聚糖G-10凝胶色谱法不适宜连续自动化分析,采用自身对照法定量时部分品种出现缔合物峰不稳定,甚至不缔合等问题,虽然每版《中华人民共和国药典》都针对暴露出的问题进行改进,但由于方法固有的缺陷,无法满足现代药品质量控制的需要已经逐渐成为专家共识[15]。这也体现在自2010年版之后,《中华人民共和国药典》未再增加利用Sephadex G-10凝胶色谱法的控制标准。HPSEC方法与葡聚糖凝胶G-10色谱法相比,虽然基本原理相同。但是HPSEC法的吸附和分子筛效应更强,且分析时间缩短,检验效率得到大幅度提升。试验结果表明,HPSEC方法分离头孢唑肟钠和聚合物杂质较为理想,也能够满足质量分析的准确度要求,可用于检测头孢唑肟钠中的聚合物等杂质。采用Sephadex G-10凝胶色谱法和 TSK G2000 SW高效凝胶色谱法分别测定头孢唑肟钠的聚合物含量,发现二者具有较明显的相关性,但TSK结果比葡聚糖凝胶G10结果高1.6倍。另外,如何确定β-内酰胺类抗生素聚合物的质控限度问题仍没有明确的答案[16]。
