冷诱导RNA结合蛋白在体外循环期间导致急性肾损伤机制实验研究
2022-08-17刘展会豆涛涛张西安
范 阳,刘展会,豆涛涛,张西安
(1.西安医学院,陕西 西安 710021;2.西安市第九医院,陕西 西安 710054)
大多数心脏手术都离不开体外循环(Cardiopulmonary bypass,CPB)。然而,在CPB 过程中,白细胞介素(Interleukin,IL)、肿瘤坏死因子-α(Tumor necrosis factor,TNF-α)、激肽释放酶和缓激肽等炎症因子的表达显著升高[1],这会加剧CPB过程中的炎性反应并导致严重的并发症[2-3]。其中,急性肾损伤(AKI)与其密切相关[4-6],也是最严重、最常见的并发症之一。从临床数据来看,心血管手术患者中5%~30%患有AKI,这些患者的病死率明显升高。同时,也给社会带来了严重的公共卫生负担和巨大的公共资源消耗。
冷诱导RNA 结合蛋白(Cold-inducible RNA-binding protein,CIRP)属于冷休克蛋白家族,是在哺乳动物细胞中发现的第一个冷休克蛋白。它在多种组织中以低水平表达,但在低温和缺氧等环境中显着上调[7-8]。在缺血再灌注损伤中,CIRP蛋白可以从细胞核转移到细胞质中,并逐渐释放到细胞外。细胞外的CIRP可以作为一种新的损伤相关模式分子(Damage-associated molecular pattern,DAMP)来激活Kupffer细胞并促进炎性反应[9]。CPB期间的心肌缺血再灌注可诱导活性氧(Reactive oxygen species,ROS)的产生,而且肾脏的线粒体含量仅次于心脏,是线粒体含量第二丰富的脏器,这可能通过上调促炎转录因子引起炎症并增加AKI的风险[10-12]。Liu等[13]报道CPB患者CIRP水平显著升高,是术后AKI的独立危险因素。另有研究发现,分泌型CIRP主要通过识别 TLR4-MD2复合物来产生炎症因子和趋化因子,从而激活细胞内炎症信号通路,进而引起非特异性炎性反应。因此,我们推测在心脏骤停期间可能会分泌大量CIRP,然后在再灌注后释放到循环中。这些分泌的CIRP可以作为DAMP诱导全身炎症并促进AKI的发展。
1 材料与方法
1.1 实验材料
1.1.1 实验细胞:大鼠心肌细胞系H9c2(批号:CL-0089),大鼠肾小管上皮细胞NRK-52E(批号:CL-0174)。
1.1.2 药品及试剂:BCA蛋白试测定剂盒(批号:P0012S);4%多聚甲醛(批号:441244);DMEM(批号:S9020);胎牛血清(批号:T8150);胰蛋白酶(批号:31600);C23(批号:BP022069);发光液(货号:1705061);冷诱导RNA 结合蛋白抗体(anti-CIRP,批号:ab191885);缺氧诱导因子1α抗体(anti-HIF-1α,批号:ab179483);TOLL样受体4抗体(anti-TLR-4,批号:ab22048)、髓样分化初级应答基因-88抗体(anti-MyD88,批号:ab133739)、半胱氨酸蛋白酶-3抗体(anti-caspase-3,批号:ab32351)、甘油醛-3-磷酸脱氢酶抗体(anti-GAPDH,批号:ab8245)、DAPI染色试剂(批号:ab104139);凋亡蛋白bax抗体(anti-bax,批号: ab216494)、凋亡蛋白bcl2抗体(anti-bcl2,批号:ab32124);TUNEL细胞凋亡检测试剂盒(批号:CASP3C);0.5% Triton X-100 (批号:0219485483),1%牛血清白蛋白 (批号:36104ES25);CIRP检测试剂盒(批号:SEG886Hu);IL-6检测试剂盒(批号:SEA079Ga);TNF-α检测试剂盒(批号:SEA133Si)。
1.1.3 主要仪器:超低温冰箱、细胞培养箱(美国Thermo Scientific公司);超净工作台(北京东联哈尔仪器制造有限公司);电子天平(瑞士Mettler Toledo公司);紫外分光光度仪(日本岛津公司);凝胶成像系统(美国Bio-Rad Laboratories公司);荧光显微镜(日本Olympus公司)。
1.2 实验方法
1.2.1 细胞培养:大鼠心肌细胞系H9c2在添加有10% FBS、100 U/ml青霉素和100 mg/L链霉素的DMEM培养基中培养。将细胞接种在6孔板中,每孔密度为5×105个细胞。将细胞分为以下四组:常温(37 ℃)常氧培养2 h(NN组),低温(15 ℃)缺氧(1% O2)培养2 h(HH组),细胞在低温和低氧条件下培养2 h,然后在常温和常氧条件下培养30 min(HHNN组),在低温和低氧条件下培养2 h的细胞,加入C23,一种CIRP衍生肽,可阻断CIRP与其受体结合,然后在常温常氧下培养30 min(C23组)。收集不同处理组的培养基。然后将6孔板中密度5×105个/孔的大鼠肾小管上皮细胞NRK-52E分别与四组培养基在常温常氧下培养4 h。
1.2.2 蛋白质印迹:通过10%十二烷基硫酸钠-聚丙烯酰胺凝胶电泳对来自每组的等量蛋白质进行电泳并转移到PVDF膜上。将膜与一抗在4 ℃孵育过夜,然后加入辣根过氧化物酶偶联的二抗,并在37 ℃下孵育1 h。本研究中使用的抗体是 anti-CIRP、anti-HIF-1α、anti-TLR-4、anti-MyD88、anti-Caspase-3 (1∶1000)、anti-bax、anti-bcl2 (1∶1000),anti-GAPDH(1∶3000)。使用发光液检测蛋白质并通过成像系统观察。
1.2.3 免疫荧光:免疫荧光染色,H9c2细胞在12孔板中生长到80%左右汇合,根据分组进行不同的处理。细胞用PBS洗涤3次,用4%多聚甲醛固定,用0.5% Triton X-100渗透。细胞用1%牛血清白蛋白封闭,并用anti-CIRP(1∶100) 在4 ℃过夜。然后将样品与二抗在37 ℃下孵育1 h,并用DAPI复染。C23被绿色荧光蛋白标记,进入细胞后即可表达,使用荧光显微镜对细胞进行成像。
1.2.4 TUNEL染色:为了在孵育后检测凋亡的NRK-52E细胞,按照制造商的方案进行了TUNEL染色。完成所有程序后,用DAPI对标本进行复染。与没有DNA片段化而显示蓝色核染色的细胞形成对比,当染色为红色时鉴定为凋亡细胞核。TUNEL测定是通过对每个样本5个图像上的3个独立样本的细胞计数来量化,然后使用单个样本的算术平均值进行统计评估。
1.2.5 酶联免疫吸附试验(ELISA):按分组进行不同处理后,收集H9c2和NRK-52E细胞培养基作为待测标本。使用ELISA 试剂盒测定 CIRP、IL-6、TNF-α的水平。所有测定均根据制造商的说明进行。实验独立重复3次。H9c2细胞培养后与NRK-52E细胞培养后IL-6和TNF-α浓度的差异计算为ΔIL-6和ΔTNF-α。
2 结 果
2.1 低温缺氧促进心肌细胞分泌CIRP 为模拟心脏手术中心肌缺血再灌注的过程,将H9c2细胞在15 ℃、1% O2条件下培养2 h,然后在常温常氧条件下培养30 min。结果表明HH组和HHNN组HIF-1α的表达高于NN组。HHNN组CIRP的表达也显著高于NN组,而CIRP在C23组和NN组的表达相似。TLR-4和MyD88的表达趋势与CIRP一致(图1A)。免疫荧光染色显示,NN组、HH组和HHNN组CIRP表达依次升高,但C23组CIRP表达明显低于HH组和HHNN组(图1B)。

A:各组CIRP、TLR-4和MyD88蛋白表达情况;B:各组免疫荧光染色(×400)
2.2 低温缺氧促进心肌细胞炎性因子分泌 低温和缺氧不仅促进心肌细胞分泌CIRP,而且促进炎症因子的分泌。各组培养基中CIRP浓度分别为(104.9±23.8) pg/ml、(170.8±20.9) pg/ml、(245.7±33.5) pg/ml和(146.7±26.6) pg/ml(HHNN与NN,P=0.006,HHNN与C23,P=0.018)。此外,各组培养基中IL-6浓度分别为(20.3±2.1) pg/ml、(33.0±1.9) pg/ml、(47.2±4.0) pg/ml和(26.3±2.3) pg/ml(HHNN与NN,P=0.002;HHNN与C23,P=0.003)。各组TNF-α浓度分别为(16.4±3.0) pg/ml、(46.5±4.1) pg/ml、(70.7±5.1) pg/ml和(35.1±3.6) pg/ml(HHNN与NN,P<0.001;HHNN与C23,P=0.001)。
2.3 低温缺氧心肌细胞分泌CIRP和炎症因子诱导肾小管上皮细胞凋亡 CPB期间,心脏再灌注后释放大量CIRP等炎症因子。使用低温缺氧的H9c2细胞培养基培养NRK-52E细胞,模拟CPB后的肾损伤。TUNEL染色表明HH组和HHNN组的凋亡细胞多于 NN 组和 C23 组(图2A)。四组的凋亡指数分别为0.92%、5.39%、8.36%和1.97%(HHNN与NN,P=0.013;HHNN与C23,P=0.009;HH与NN,P=0.016,图2B)。此外,凋亡基因 cleaved caspase-3和bax在HH组和HHNN组中的表达高于NN组和C23组,而凋亡抑制基因bcl-2在HH组和HHNN组中低表达(图2C)。另外CIRP还刺激了NRK-52E细胞的炎性反应。HHNN组TLR-4和MyD88的表达高于NN组和C23组。各组NRK-52细胞分泌的ΔTNF-α浓度分别为(4.9±0.2) pg/ml、(16.5±4.0) pg/ml、(15.7±3.6) pg/ml和(8.8±2.2) pg/ml(HHNN与NN,P=0.034,HH与NN,P=0.038,图2D)。各组ΔIL-6浓度分别为(8.4±1.5) pg/ml、(9.4±3.0) pg/ml、(12.1±0.7) pg/ml和(7.2±1.0) pg/ml(HHNN与NN,P=0.032;HHNN与C23,P=0.004,图2E)。
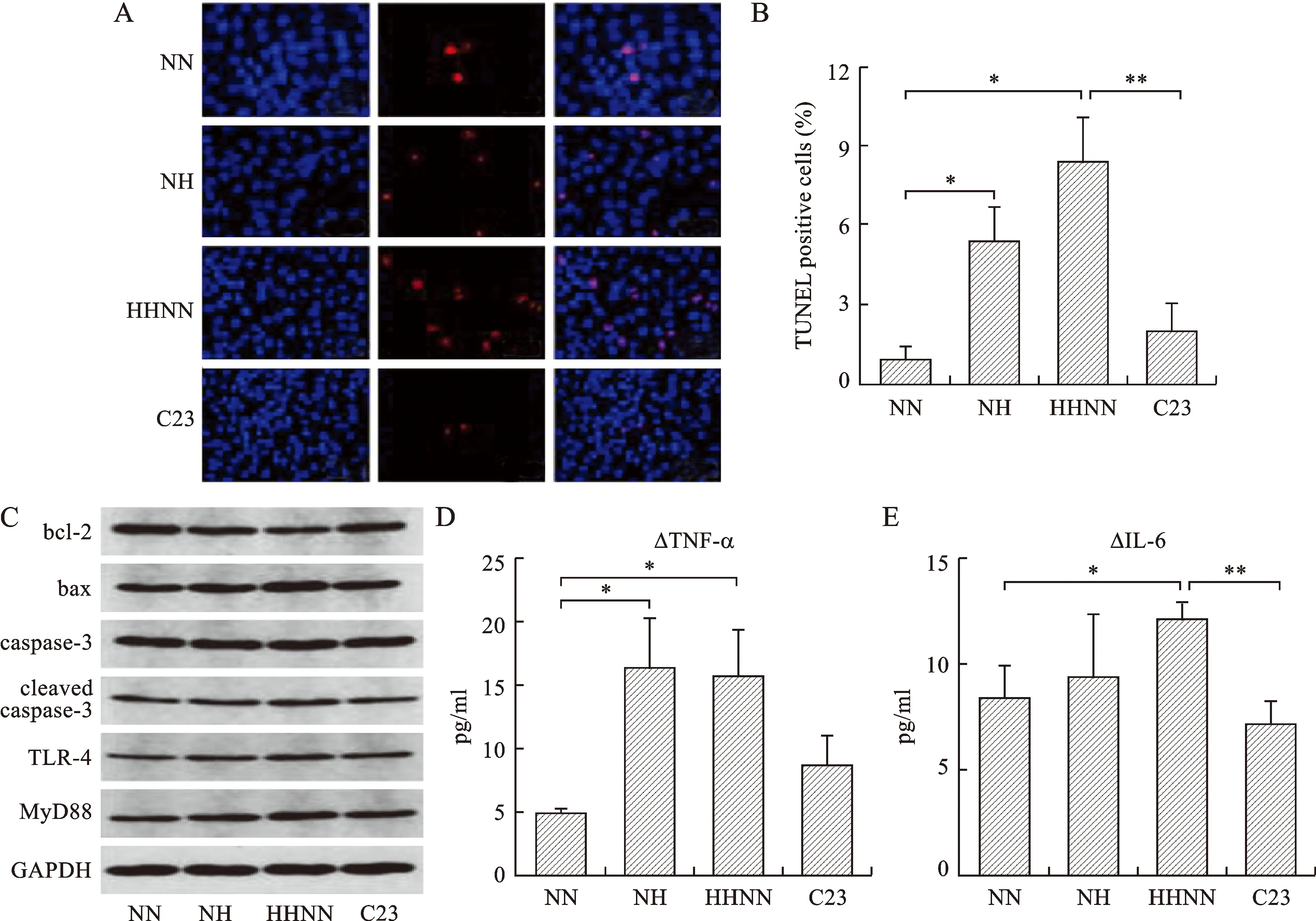
A:各组TUNEL染色(×200);B:各组细胞凋亡指数;C:各组相关因子蛋白表达情况;D、E:各组培养基中△TNF-α、△IL-6浓度。两者比较,*P<0.05,**P<0.01
3 讨 论
已知细胞内CIRP和细胞外CIRP具有不同的功能,细胞内CIRP充当RNA伴侣以协调与应激源相关的翻译,而细胞外CIRP充当新的DAMP以促进和放大炎症级联[13]。在许多以无菌炎症为特征的缺血再灌注模型中,CIRP的血清和组织水平也显著升高。Zhou等[14]发现,大脑中动脉闭塞后小鼠脑中CIRP升高,并随着缺氧时间增加分泌到细胞外,同时TNF-α表达上调,而CIRP缺陷小鼠的同一模型中TNF-α的表达和小胶质细胞的活化显著降低。Cen等[15]研究表明,CIRP的缺乏可以减轻炎症和氧化应激,并减轻肾缺血再灌注后的肾损伤。
先前的研究表明,CIRP 与参与NF-κB激活的细胞表面受体TLR4-MD2复合物具有强结合力,激活细胞内炎症信号通路产生炎症因子与趋化因子,进而引起非特异性炎症反应[13],而 TLR4缺陷小鼠对重组 CIRP 蛋白刺激诱导的炎性反应显著弱于野生型小鼠。在组织骨折模型中,CIRP 可通过TLR4/MyD88 通路激活 NADPH 氧化酶引起 ROS 释放,并活化核酸内切酶 G 致使线粒体碎片化,最终介导巨噬细胞自噬与程序性坏死[16]。在本研究中,我们发现心肌细胞在低温缺氧的情况下会分泌大量的CIRP。一方面,这些CIRP激活心肌细胞的炎症反应,并通过TLR-4/MyD88途径释放TNF-α和IL-6。另一方面,分泌的CIRP等炎症因子作用于肾小管上皮细胞,诱导其凋亡。C23与CIRP 序列全长的15聚体肽重叠,对MD2具有高亲和力。它阻断了CIRP介导的促炎反应,减少了对肾小管上皮细胞的损伤。
HIF-1在缺氧环境中的作用已被广泛研究。大量信号分子由HIF-1 DNA结合转录诱导,从而增加细胞氧合并增强对缺氧条件的代谢适应。目前关于缺氧条件下HIF-1α与CIRP关系的研究较少。Wellmann证明CIRP通过一种不依赖HIF-1的机制在缺氧时适应性地表达。Chen等[17]发现,在体内和体外慢性低压缺氧条件下,HIF-1α均显著升高,并介导神经元凋亡。然而,CIRP 的过表达抑制了缺氧时 HIF-1α的上调,并抑制了缺氧诱导的神经元凋亡。在我们的研究中,CIRP 和 HIF-1α 的表达在暴露于低温和缺氧的心肌细胞中增加,并且在C23给药后HIF-1α和CIRP的表达受到抑制。在癌症研究中也有类似的结果。Lu等[18]报道CIRP可以通过结合其 mRNA 的3’-UTR来诱导HIF-1α的表达,从而增加膀胱癌细胞中 mRNA 的稳定性。Yang等[19]还证明HIF-1α是Uni Gene 3’-UTR数据库中 CIRP 靶向转录物之一。有趣的是,CIRP 的表达在复温和复氧后继续增加。在大鼠体外循环模型中发现了类似的结果。CIRP的表达在心跳恢复后的复温阶段继续增加[20]。相关机制还有待进一步研究。
综上所述,心脏手术过程中,心肌细胞不可避免地需要经历低温缺氧的过程。我们的研究结果表明心肌细胞在低温和缺氧条件下分泌大量CIRP,并在复温和复氧后诱导炎症因子的产生。分泌的CIRP可介导炎症级联反应并促进肾小管上皮细胞凋亡,进一步的结论需要更多的动物实验结果来证明。
