马波沙星原料质量标准的建立
2022-08-11董玲玲龚旭昊赵富华
董玲玲,张 璐,杨 星,龚旭昊,赵富华
(中国兽医药品监察所,北京100081)
马波沙星(Marbofloxacin),又叫麻保沙星,最早由瑞士罗氏公司研制,并于1995年先后在英法等国上市[1],国内兽药生产企业自2014年起陆续申报马波沙星原料及其制剂。马波沙星是兽医专用的第三代喹诺酮类抗菌药物,具有抗菌谱广,消除半衰期长,生物利用度高的特点,兽药市场发展前景良好,但是国内缺乏统一的质控标准。为进一步加强对兽药安全性和有效性的控制,提高兽药产品质量,将研究制定马波沙星原料质量标准纳入《中国兽药典》2020年版科研任务。国内能查到马波沙星原料相关质量标准5个,为申报企业的注册标准[2-6],欧洲药典(EP 9.0)和英国药典(BP 2017)也收载了马波沙星原料质量标准[7],对各质量标准进行比较,欧洲药典和英国药典收载的质量标准相同,所有标准中吸光度、干燥失重、炽灼残渣、重金属、含量测定方法相同,其余项目存在差异。在文献查阅的基础上,着重研究有关物质、残留溶剂检查和含量测定方法。最终建立的马波沙星原料质量标准收载入《中国兽药典》2020年版一部[8],内容包括:性状、鉴别(紫外光谱法和红外光谱法)、吸光度、有关物质、干燥失重、炽灼残渣、重金属、含量测定等项目。
1 仪器与试药
高效液相色谱仪(Waters e2489)配二极管阵列检测器(2996);瑞士万通全自动滴定仪Titrando 809; AT201分析天平(Mettler Toledo); 色谱柱: Waters XTERRA RP18(4.6×150 mm,5 μm); Waters XBridge C18(4.6×150 mm, 5 μm); GRACE C18(4.6×150 mm,3 μm)。甲醇为色谱纯,购自Merck公司。辛烷磺酸钠、磷酸二氢钠均为分析纯,购自TCI公司。其余试剂均为分析纯。马波沙星原料6批,分别购自浙江国邦药业股份有限公司(批号:160919;161011;170306)和海门慧聚药业有限公司(批号:171101;171201;171202)。马波沙星对照品,中国兽医药品监察所,批号:H0801511;马波沙星杂质峰鉴别对照品,EP,批号:Y0002317-1。
2 方法与结果
2.1 有关物质检查方法
2.1.1 方法的建立过程
2.1.1.1 色谱柱的选择 现有标准方法流动相基本一致,仅比例略有不同,因此,选用EP方法中的流动相条件,并在此基础上进行色谱柱的选择。现有标准方法中色谱柱填料类型均为十八烷基硅烷键合硅胶。研究过程中发现使用不同品牌C18色谱柱,马波沙星主峰及各杂质峰保留时间差别较大,且各杂质相对于主峰的保留时间会发生变化,试验三种不同品牌色谱柱,主峰保留时间及各杂质相对主峰保留时间结果见表1。由表1可知,不同品牌粒径的C18色谱柱对马波沙星主峰及各杂质峰的保留时间有影响。为保证主峰时间不至于过长,且有稳定的相对保留时间,因此在质量标准中提供色谱柱品牌型号信息作为参考,拟定色谱柱条件为“用十八烷基硅烷键合硅胶为填充剂(XTERRA C18,4.6×150 mm,5 μm或其他效能相当的色谱柱)”。
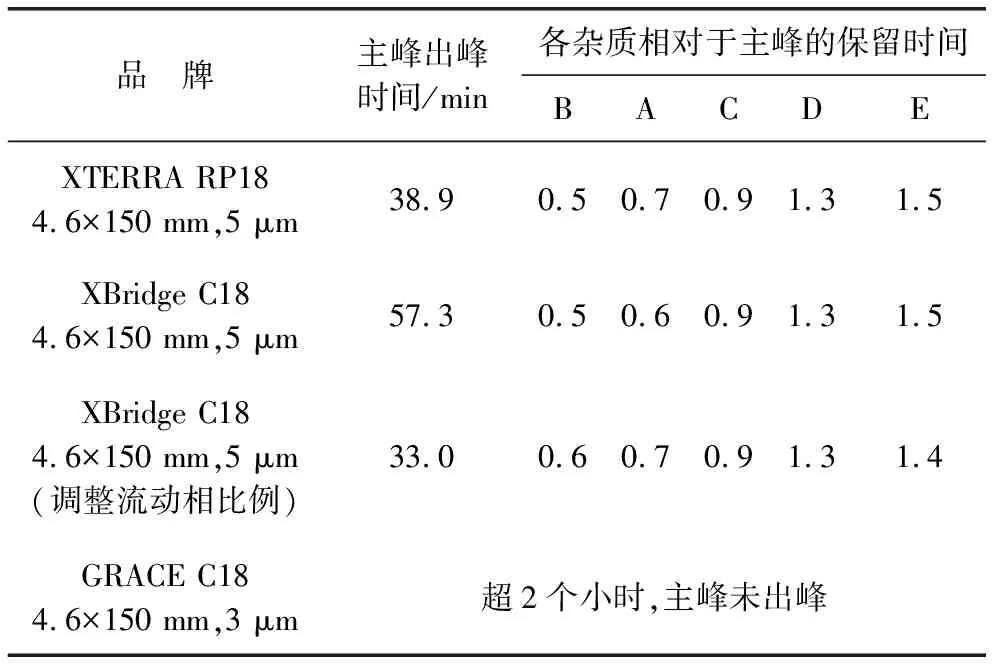
表1 不同品牌色谱柱马波沙星主峰保留时间及各杂质相对主峰保留时间结果表
2.1.1.2 各杂质的相对保留时间的确定 杂质鉴别对照品是一支混合对照品,溶解进样后发现在主峰之前有四个色谱峰,相对主峰的保留时间分别为0.5,0.7,0.8和0.9,给杂质A和C的定位造成干扰。只能单独进样杂质A和C的定位溶液,确定了杂质A和C相对于马波沙星主峰的保留时间分别为0.7和0.9(图1)。取杂质鉴别对照品适量,加甲醇-水(23∶77)稀释制成每1 mL中约含1 mg的溶液作为系统适用性溶液,精密量取10 μL,按拟定液相色谱条件(2.1.2项)连续进样6针,主峰及各杂质峰的保留时间略有漂移,但各杂质峰相对马波沙星峰的保留时间不变,其结果见表2。综上,拟定杂质B、A、C、D、E的相对保留时间分别为0.5,0.7,0.9,1.3,1.5。
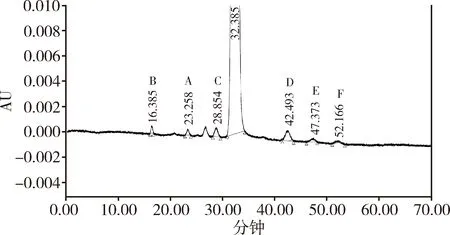
图1 马波沙星系统适用性溶液图

表2 相对保留时间结果表
2.1.1.3 杂质限度规定 马波沙星的6个杂质中,杂质C和D为合成中间体,杂质F主要在长期贮存过程产生。国内外各质量标准中各杂质限度及总杂质限度均相同,仅忽略限有不同。结合6批马波沙星原料的有关物质检查结果,各杂质限度直接参考EP的设定。目前未对杂质F作特别的规定,计入总杂质达到控制的目的,在积累稳定性数据之后,再考虑对其限度进行规定。EP标准中将杂质忽略限设定为0.1%,与杂质A、B的限度一致,杂质A、B只能是未检出和不符合规定两种结果,欠合理。结合方法检测限和灵敏度考察结果,将忽略限定为0.05%。
2.1.2 有关物质质量标准 取本品适量,加甲醇-水(23∶77)适量,超声处理使溶解并稀释制成每1 mL中约含1 mg的溶液,作为供试品溶液;精密量取适量,加甲醇-水(23∶77)稀释制成每1 mL中约含1 μg的溶液作为对照溶液。照高效液相色谱法(附录0512)测定,用十八烷基硅烷键合硅胶为填充剂(XTERRA C18,4.6×150 mm,5 μm或效能相当的色谱柱)。以甲醇-冰醋酸-磷酸盐缓冲液(取磷酸二氢钠2.7 g、辛烷磺酸钠3.5 g,加水1000 mL使溶解,用磷酸调节pH值至2.5)(230∶5∶770)为流动相;检测波长315 nm;流速为1.2 mL/min;柱温为40 ℃。取马波沙星杂质峰鉴别对照品(含杂质A、B、C、D、E)10 mg,置10 mL量瓶中,加甲醇-水(23∶77)稀释至刻度,摇匀,作为系统适用性溶液,精密量取10 μL注入液相色谱仪,记录色谱图至主成分峰保留时间的2.5倍,马波沙星峰保留时间约为33 min,杂质B、A、C、D、E峰相对于马波沙星峰的保留时间分别约为0.5、0.7、0.9、1.3和1.5,杂质C峰和马波沙星峰的分离度应大于1.5,杂质D峰和马波沙星峰的分离度应大于4.0。精密量取供试品溶液和对照溶液各10 μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的2.5倍。供试品溶液色谱图中如有杂质峰,杂质A、B峰面积不得大于对照溶液主峰面积(0.1%);杂质C、D峰面积不得大于对照溶液主峰面积的2倍(0.2%);杂质E按校正后峰面积计算(乘以校正因子1.5)不得大于对照溶液主峰面积的2倍(0.2%);按校正后峰面积计算各杂质峰面积之和不得大于对照溶液主峰面积的5倍(0.5%)。供试品溶液色谱图中小于对照溶液主峰面积0.5倍(0.05%)的色谱峰忽略不计。
2.1.3方法学验证
2.1.3.1 专属性 溶剂空白:以甲醇-水(23∶77)作为溶剂空白。系统适用性溶液:取马波沙星杂质峰鉴别对照品(含杂质A、B、C、D、E)10 mg,置10 mL量瓶中,加甲醇-水(23∶77)稀释至刻度,摇匀,过滤,取续滤液,即得。供试品溶液:取供试品10 mg,置10 mL量瓶中,加甲醇-水(23∶77)稀释至刻度,摇匀,过滤,取续滤液,即得。
精密量取上述溶液各10 μL,按照拟定质量标准有关物质色谱条件进行测定,结果显示在溶剂空白图谱中(图2),马波沙星主峰及各杂质峰出峰处无干扰;杂质鉴别对照品色谱图中,马波沙星主峰及各杂质色谱峰峰纯度均小于阈值,均为单一物质峰。
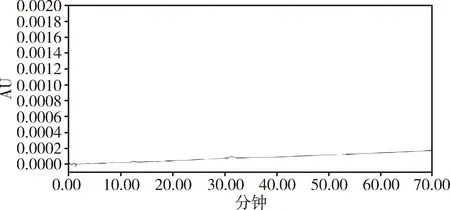
图2 马波沙星有关物质溶剂空白色谱图
2.1.3.2 检测限 称取马波沙星对照品10 mg,置10 mL量瓶中,加甲醇-水(23∶77)适量,超声使溶解,并稀释至刻度,摇匀,精密量取适量0.1 mL至100 mL量瓶中,得0.1%(1 μg/mL)的溶液,再精密量取适量,加甲醇-水(23∶77)系列稀释,得0.05%(0.5 μg/mL)、0.02%(0.2 μg/mL)、0.01%(0.1 μg/mL)的溶液,精密量取各溶液10 μL,按照拟定的有关物质检查的色谱条件进行测定,结果显示,0.01%溶液主峰信噪比为4.3,故有关物质测定法的检测限为0.01%。
2.1.3.3 精密度(重复性) 配制浓度为0.1%(1 μg/mL)的马波沙星对照品溶液和系统适用性溶液,精密量取上述两种溶液各10 μL,按照拟定标准中有关物质色谱条件进样测定,平行测定6次。结果:在0.1%的浓度水平下,6次测定马波沙星峰面积RSD为1.1%;系统适用性溶液6次测定下各杂质相对于马波沙星主峰的保留时间不变,该方法的精密度良好。
2.1.3.4 耐用性 既定流速为1.2 mL/min,柱温为40 ℃。调节流速为1.0、1.3 mL/min,调节柱温为38、42 ℃,以系统适用性溶液按照拟定标准中有关物质色谱条件进样测定,考察方法的耐用性,结果当流速在1.0~1.3 mL/min的范围内变化,柱温在40±2 ℃的范围内变化,检出的杂质峰个数未发生改变,各杂质峰之间的分离情况符合规定,该方法的适用性良好。
2.1.3.5 溶液稳定性 取一批供试品溶液,按照拟定色谱条件,分别在第0、4、8、12、24 h进样测定,考察检出杂质峰个数和杂质含量,结果在24 h内,检出的杂质峰个数不变,杂质量及总杂质量未发生改变,供试品溶液稳定。
2.1.4 有关物质检查结果 取6批马波沙星原料,按拟定色谱条件检验,结果见表3。所得结果与生产企业自检报告中结果一致,方法可行。
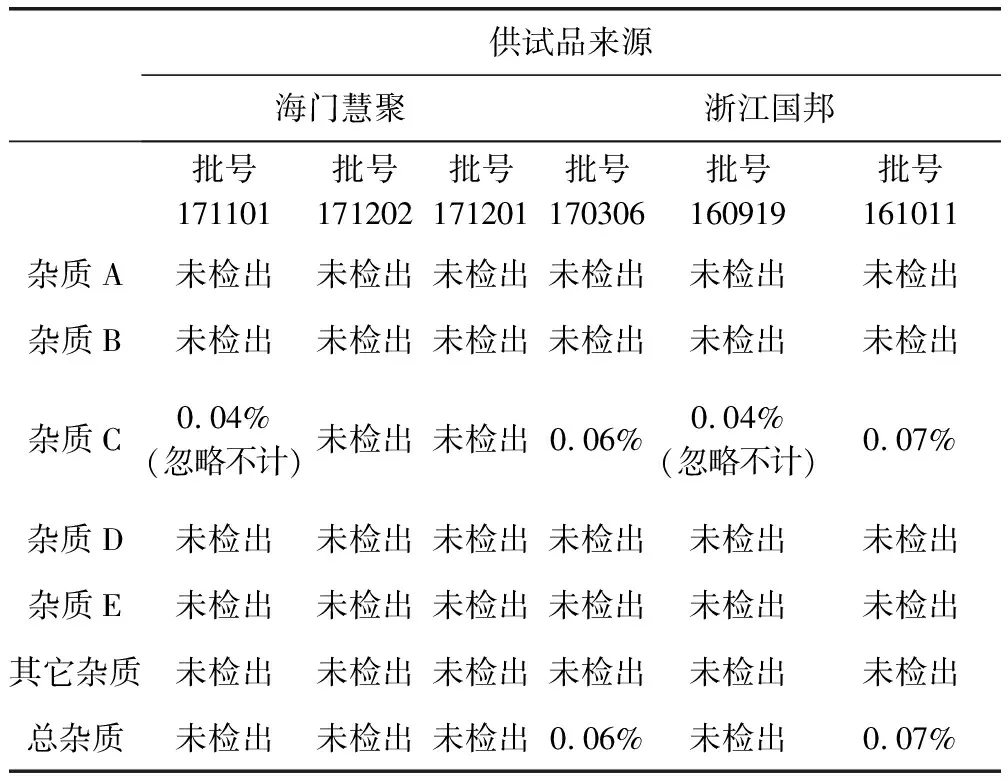
表3 马波沙星原料有关物质检查结果表
2.2 残留溶剂检查方法
2.2.1 残留溶剂的检查方法建立与方法学考察 见作者发表的文章:顶空毛细管GC法测定马波沙星原料药中6种溶剂的残留量[9]。
2.2.2 残留溶剂质量标准 取供试品约0.1 g,精密称定,置顶空瓶中,精密加入N,N-二甲基甲酰胺1 mL,密封,作为供试品溶液;分别精密称取甲醇、乙醇、乙腈、二氯甲烷、苯与甲苯适量,加N,N-二甲基甲酰胺定量稀释制成每1 mL中分别含甲醇0.3 mg、乙醇0.5 mg、乙腈0.041 mg、二氯甲烷0.06 mg、苯0.002 mg和甲苯0.089 mg 的混合溶液,精密量取1 mL,置顶空瓶中,密封,作为对照品溶液。照残留溶剂测定法(附录0861第二法)测定。以6%氰丙基苯基-94%二甲基聚硅氧烷(或极性相近)为固定液的毛细管柱为色谱柱;初始温度为40 ℃,维持10 min后,以每分钟20 ℃的速率升温至80 ℃,维持2 min,再以每分钟30 ℃的速率升温至200 ℃,维持5 min。以氮气为载气,线速为每秒24.1 cm,分流比为5∶1;进样口温度为180 ℃;检测器为火焰离子化检测器(FID),检测器温度为250 ℃;顶空瓶平衡温度为105 ℃,平衡时间为20 min。取对照品溶液顶空进样,记录色谱图,出峰顺序依次为:乙醇、乙腈、二氯甲烷、苯与甲苯,各色谱峰间的分离度应符合要求。取供试品溶液和对照品溶液分别顶空进样,记录色谱图,按外标法以峰面积计算,均应符合规定。
2.2.3 残留溶剂检查结果 取6批马波沙星原料,按拟定残留溶剂检查方法检验,结果见表4。所得结果与生产企业自检报告中结果一致,方法可行。

表4 马波沙星残留溶剂结果表
2.3 含量测定方法
2.3.1 含量测定方法的确立 国内外现有质量标准中含量测定方法相同,均为电位滴定法。考虑到原料纯度高且不存在辅料的干扰,容量法测定含量专属性良好。为进一步验证容量法测定含量的可靠性,参考马波沙星制剂的含量测定方法(HPLC法,外标法)[2]测定6批马波沙星原料的含量,与电位滴定法测定结果相比较(表5),相对偏差不大于0.02%,最终仍以电位滴定法作为马波沙星含量测定方法。
2.3.2 含量测定方法 取本品约0.3 g,精密称定,加冰醋酸80 mL使溶解,照电位滴定法(附录0701),用高氯酸滴定液(0.1 mol/L)滴定,并将滴定的结果用空白试验校正,每1 mL盐酸滴定液(0.1 mol/L)相当于36.24 mg的C17H19FN4O4。
2.3.3 含量测定结果 取6批马波沙星原料,按拟定含量测定方法检验,结果见表5。
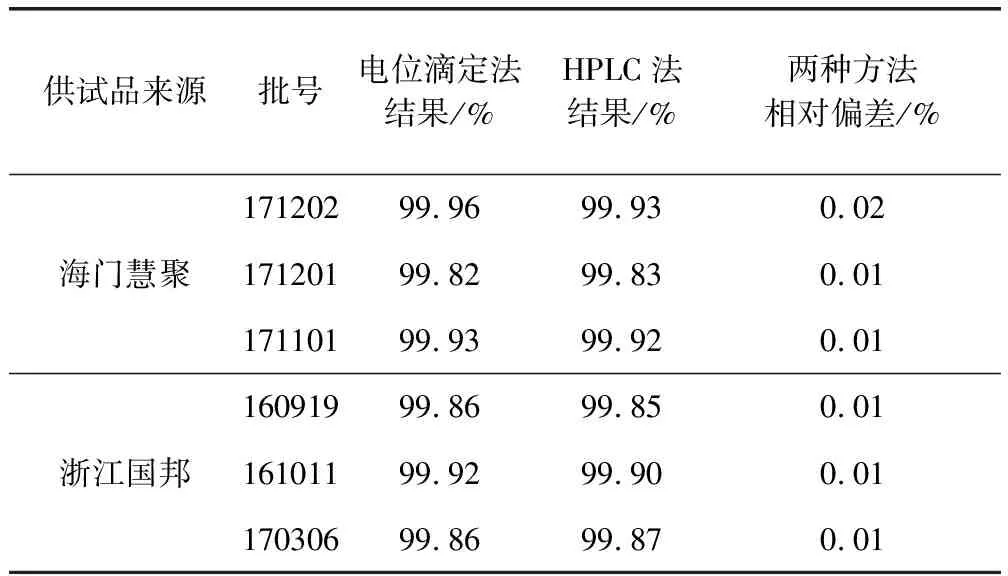
表5 马波沙星原料含量测定结果表
3 讨 论
3.1 破坏试验 为进一步验证有关物质检查方法对杂质的检出能力,研究过程中进行破坏试验,采用强光照射,酸或碱破坏,氧化破坏及加热等方式。按照拟定的有关物质色谱条件进行测定,马波沙星原料在光照、加热和常规酸碱破坏条件下比较稳定。又采用酸碱破坏同时水浴的办法,破坏产生杂质,主要为杂质D、E、F,均为方法本身控制的杂质。方法本身控制的杂质种类齐全,已包括合成中间体杂质,说明方法检出能力强。
3.2 质量标准中未收录残留溶剂检查的原因 因各企业生产工艺不同,残留溶剂种类也不尽相同,虽然已建立的残留溶剂质量标准涵盖了所有可能出现的残留溶剂种类,但参考各生产企业残留溶剂检查结果,仅检出乙醇和甲苯两种溶剂残留,且残留量均远低于限量,因此最终建立的质量标准中未收录残留溶剂检查项。
3.3 质量标准中的其他项目 除了对有关物质、残留溶剂和含量测定项目进行了重点研究之外,其余项目均参照EP质量标准,采用6批马波沙星原料进行验证。最终建立的马波沙星原料质量标准包括性状、鉴别(紫外光谱法和红外光谱法)、吸光度、有关物质、干燥失重、炽灼残渣、重金属、含量测定等项目,内容完善,可用于马波沙星原料的质量控制,收载入《中国兽药典》2020年版一部。
