Core-Shell Au@NiS1+x Cocatalyst for Excellent TiO2 Photocatalytic H2 Production
2022-08-11WagehAhmedAlGhamdiQuanlongXu
S. Wageh , Ahmed A. Al-Ghamdi , Quanlong Xu
1 Department of Physics, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia.
2 Key laboratory of Carbon Materials of Zhejiang Province, College of Chemistry and Materials Engineering, Wenzhou University,Wenzhou 325027, Zhejiang Province, China.

Fig. 1 Schematic illustration of charge transfer within TiO2/Au@NiS1+x: (a) before contact, (b) contact in dark, and(c) under UV-light illumination: the (1) photoelectron trapping by the Au mediator, (2) photoelectron transfer from Au to NiS1+x, and(3) fast H2 evolution on the electron-enriched Sδ- active sites of Au@NiS1+x cocatalyst.Reproduced with permission from Ref. 8. Copyright 2022, Wiley.
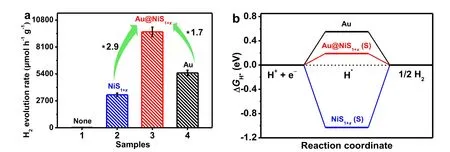
Fig. 2 (a) Photocatalytic H2-evolution rate over (1) TiO2, (2) TiO2/NiS1+x, (3) TiO2/Au@NiS1+x(1.7 : 1.3), and (4) TiO2/Au.Reproduced with permission from Ref. 8. Copyright 2022, Wiley.
The convertion of solar energy into hydrogen energy with high energy density by photocatalysis is a green and eco-friendly avenue to solve the energy crisis and environmental pollution issues1,2. Unfortunately, most photocatalysts usually suffer from the rapid quenching of photogenerated carriers and depressed interfacial H2-generation dynamics3,4. To overcome the above shortcomings, cocatalysts are widely employed to promote the separation of photocarriers and to create active sites for surface catalytic reactions5. For a superb cocatalyst, its active sites usually play a key role in the overall performance of photocatalytic H2evolution because the active sites can not only provide a large number of adsorption centers to enrich H+, but also present an outstanding catalytic efficiency to convert H+into H2by reducing its reaction overpotential6. Thus, the activesite number and efficiency of cocatalysts should be maximized as much as possible to greatly boost the cocatalytic H2-evolution activity by photocatalysis.
In recent decades, metal sulfides have been widely researched as effective cocatalysts to replace Pt-group metals because of their excellent charge capture capability and cost-effective property. It is commonly accepted that the S atoms on the sulfide cocatalysts can act as the active sites to capture and enrich protons (H+) from solution, and enhance the final H2formation7.However, the H2-evolution activities of sulfide cocatalysts are usually limited by insufficient quantities of exposed sulfur (S)sites and their strong bonding with adsorbed hydrogen atoms (SHads, 363 kJ·mol-1) compared with that of Pt-Hads(251 kJ·mol-1,the most efficient cocatalyst) (Fig. 1a)8.
To address these issues, an innovative strategy of active-siteenriched regulation and electronic structure modification of active S sites has been firstly developed by Yu and co-workers(China University of Geosciences), with the aim of simultaneously increasing the active S number and weakening the strong S-Hadsbonds (Fig. 1) (Advanced Materials2022,34,2108475. doi: 10.1002/adma.202108475)8. In this case, they rationally designed a core-shell Au@NiS1+xnanostructured cocatalyst by a two-step route, including initial photodeposition of Au nanoparticles onto the TiO2surface and the subsequent self-assembling of sulfur-enriched amorphous NiS1+xon the Au surface by a selective adsorption-in situphotodeposition method.Photocatalytic experiments exhibited that the resulting TiO2/Au@NiS1+x(1.7 : 1.3) displayed a boosted H2-generation rate (9616 μmol·h-1·g-1, AQE = 46.0%,λ= 365 nm), which is 2.9 and 1.7 times over the TiO2/NiS1+xand TiO2/Au, respectively(Fig. 2a). Encouragingly, large amount of visual H2bubbles can be observed under UV-light irradiation by using TiO2/Au@NiS1+x(1.7 : 1.3) as photocatalyst. In addition, the authors proved that the designed Au@NiS1+xcan be used as a general cocatalyst for greatly boosting the H2-evolution activity of various photocatalysts (such as g-C3N4and CdS). More importantly, the innovative strategy was also applied for the fabrication of other core-shell cocatalysts with enriched active S species (such as core-shell Au@CoS1+x) to achieve improved H2-production activities.
Mechanistic studies reveal that the free electrons of Au can effectively transfer to sulfur-enriched NiS1+xto induce the generation of electron-enriched Sδ-active centers (Fig. 1b),which boosts the desorption of Hadsfor rapid hydrogen formationviaweakening the strong S-Hadsbonds (Fig. 1c). From theex situXPS results, the binding-energy shift towards a lower value was found for S 2pin TiO2/Au@NiS1+x, illustrating the formation of electron-enriched Sδ-atoms. Synchronously, the planaraveraged electron density difference Δρ(z) and charge density distributions were obtained by density functional theory (DFT)to qualitatively and quantitatively reveal the electron transfer between NiS1+xand Au, respectively, and the results further suggested that the introduction of Au led to electron enrichment at the Sδ-active sites in Au@NiS1+x. In addition, the effect of electron enrichment at Sδ-atoms on S-Hadsbinding strength was studied via the calculated free energy change (ΔGH*) (Fig. 2b).It is found that the Au@NiS1+xpresents a moderate ΔGH*value of 0.19 eV (close to the ideal ΔGH*value of zero) due to the reduced S-Hadsbinding strength, significantly lower than that of NiS1+x(-1.02 eV). Consequently, the Au@NiS1+xcocatalyst possesses an appropriate H adsorption ability, which allows rapid Hadsdesorption and combination to form H2. These results indicate the promotional effect of Au on the generation of electron-enriched Sδ-species and the weakening of S-Hadsbonding in sulfur-rich NiS1+x, which contributes to the increased catalytic efficiency of active S sites toward H2evolution. In this case, the authors rationalize the accelerated H2-production rate over TiO2/Au@NiS1+xby an electron-enriched Sδ--mediated mechanism, namely, the massive Sδ-atoms as the active sites can not only provide sufficient proton-adsorption centers to enrich H species, but also present an outstanding catalytic efficiency to fleetly convert atomic hydrogen to H2.
The work also highlights the importance ofin situcharacterization technique for unraveling photoexcited chargetransfer pathway and process of cocatalyst-loaded photocatalysts.To this end, they appliedin situirradiated X-ray photoelectron spectroscopy (ISI-XPS) to uncover the photoexcited chargetransfer pathway among TiO2, Au and sulfur-rich NiS1+x. The results showed that the photoelectrons in TiO2were first captured by the Au mediator, and then the Au core transfers the captured photoelectrons to the NiS1+xshell. Finally,photoelectrons were accumulated on the electron-enriched Sδsites as the active centers for H+reduction to H2(Fig. 2c).
In summary, the optimizing of atomic hydrogen desorption through constructing electron-enriched Sδ-sites on sulfurenriched NiS1+xpresents an innovative strategy for boosting photocatalytic H2-evolution activity of sulfide cocatalysts. This pioneering work indicates the importance of synergistic effect by increasing active-site number and efficiency for designing robust H2-evolution cocatalyst. Inspired by this innovative study, the presented strategy is expected to be utilized to construct more advanced functional cocatalyst for energy and environmental applications, such as photocatalytic CO2reduction, N2fixation,organic synthesis, selective oxidation, organic degradation,et al.9,10. In addition, the reported work will bring new insight and inspire for further innovative study on designing and constructing highly-efficient cocatalysts.
Acknowledgments:This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant No. RG-72-130-42. The authors,therefore, acknowledge with thanks DSR for technical and financial support.
杂志排行

物理化学学报的其它文章
- Insights into Mechanism of CsPbBr3 Nanocrystal Interfacial Modifier in Perovskite Solar Cells
- Enhanced Photocatalytic CO2 Reduction over 2D/1D BiOBr0.5Cl0.5/WO3 SScheme Heterostructure
- Hollow NiCo2S4 Nanospheres as a Cocatalyst to Support ZnIn2S4 Nanosheets for Visible-Light-Driven Hydrogen Production
- A 0D/2D Bi4V2O11/g-C3N4 S-Scheme Heterojunction with Rapid Interfacial Charges Migration for Photocatalytic Antibiotic Degradation
- Rationally Designed Mn0.2Cd0.8S@CoAl LDH S-Scheme Heterojunction for Efficient Photocatalytic Hydrogen Production
- P-Doped g-C3N4 Nanosheets with Highly Dispersed Co0.2Ni1.6Fe0.2P Cocatalyst for Efficient Photocatalytic Hydrogen Evolution