Fe2MnSn合金的成相和磁性研究
2022-08-11李歌天常康康张强强柳祝红
李歌天, 常康康, 张强强, 柳祝红
(北京科技大学 数理学院, 北京 100083)
Heusler合金是一种高度有序的金属间化合物,分子式为X2YZ,其中X,Y为过渡族金属元素,Z为sp元素[1-3]。最近发现部分Heusler合金具有丰富的成相现象。Zhang等[4]采用第一性原理计算研究了二元Heusler合金Mn3Z(Z为Ga、Ge和Sn)的立方相、四方相和六角相的结构稳定性及磁性。此外,已报道了实验上Mn3Ga可形成面心立方、四方及六角结构[5-8]。不同结构的Mn3Z合金往往展现出不同的物理性质。如,四方结构的Mn3Ga展现出较大的矫顽力、较低的1.1μB(μB为波尔磁子,μB=9.274×10-24A·m2)磁矩及高达730 K的居里温度,使它在自旋转移力矩器件中具有广阔的应用前景[5,9];六角结构的Mn3Z(Z为Ga、Ge和Sn)具有非共线反铁磁结构,材料中具有大的反常霍尔电导[10-12]。同时,在Mn3Ga和Mn3Sn中观察到了拓扑霍尔效应[13-14]。Mn3Sn中掺杂部分Fe会保持六角结构[15-16]。此外,通过Fe替代部分Mn形成的Mn3-xFexGa(x= 0,0.2,0.4,0.6,0.8,1)合金也可展现出多重晶体结构[8]。继续提高Fe的掺杂量,并不会影响合金多样化的相结构,如Fe2MnGa合金也展现出多种晶体结构,包括体心立方Heusler相(B2相)[17]、面心立方相[18]和密排六方结构DO19相[17]。成分稍微偏离正分配比的Fe2MnGa的B2相在一定条件下会发生马氏体相变,转变为体心四方马氏体相[19]。但如果合金中存在几种不同相的共存,改变了母相的化学成分,马氏体相变就难以出现。文献报道,温度为500 ℃以上时,Fe2MnGe为六角结构[20],但在低温下是立方L21结构[21-22],且在球磨样品中观察到了不可逆的立方到六角结构的相变[23]。最近,研究人员在室温下获得了六角结构的Fe2MnGe[24],同时罗鸿志等[25]利用第一性原理,对Fe2MnGe的相稳定性进行了系统研究。从能量的角度分析,Fe2MnGe应以L21立方结构存在,且呈现半金属性。理论计算发现,在相同的16个原子的晶胞里,L21立方结构和六角结构之间的能量差为0.8 eV,六角结构会破坏材料的半金属性质。利用第一性原理计算发现四方结构的Fe2MnSn具有铁磁特征,居里温度高达1 012 K,磁矩为7.34μB[26]。早期有文献报道高能球磨可获得立方Heusler结构的Fe2MnSn[27],最近也有文献报道合成了六角结构的Fe2MnSn[28]。
材料的性质很大程度上取决于其结构,相同的分子式,不同的晶体结构往往表现出不同的物理性质,尤其是在易形成多相性的材料中。为发展多功能材料,有必要对它们的成相进行研究。本文从第一性原理出发,根据磁性对成相的影响规律,研究了Fe2MnSn的成相规律,解释了电弧炉熔炼很难得到立方结构Fe2MnSn的原因,并对Fe2MnSn的磁性进行了分析。
1 计算和实验方法
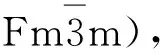
计算中,采用广义梯度近似(generalized-gradient approximation,GGA)来描述电子交换关联常数,采用对所有元素都有很高效率的超软赝势(ultrasoft pseudo potential,UPP)及500 eV的截断能量,自洽收敛能量为10-7eV。对于立方结构总能量收敛及态密度的计算采用9×9×9个k点的k网格进行布里渊区域积分。对六角结构的总能量收敛及态密度计算采用9×9×8个k点。
在合成样品时,单质金属Fe,Mn和Sn的纯度均在99.9%以上。按照名义成分Fe2.1MnSn0.9进行化学配比,然后在氩气保护下,采用电弧炉熔炼制备多晶样品。熔炼后的样品在真空石英管中进行800 ℃高温退火3 d,随后随炉冷却到室温。样品的晶体结构采用X射线衍射(X-ray diffraction,XRD)实验进行表征,所用仪器为日本理学公司的Smartlab3型X射线衍射仪。样品的磁性测量采用综合物性测量系统(physical properties measurement system,PPMS)进行测量。
2 实验结果和讨论
3种不同结构Fe2MnSn化合物的能量随体积的变化关系如图2所示。本文使用内含16个原子的不同结构的晶胞来进行比较。由图2可见:在3种结构中,XA立方结构的能量最低,其次是DO19六角结构,L21立方结构的能量较高。值得指出的是,DO19六角结构的能量与XA立方结构的最低能量非常接近,两种结构的能量差仅为0.01 eV。从能量角度分析可知,XA结构的Fe2MnSn最稳定,所以合金应以XA结构存在。按Heusler合金的优先占位规则,价电子数多的电子应优先占据A位和C位[29]。Fe原子的价电子数多于Mn原子的价电子数,按照占位规则,合金应该形成Fe-Mn-Fe-Sn占位的L21结构,而不是形成Fe-Fe-Mn-Sn型的XA结构,合金Fe2MnSn违背了Heusler合金的优先占位规则。实际上,随着Heusler合金种类的不断增加,人们发现并不是所有的Heusler都遵循优先占位规则,如Ti2CrZ(Z=Al,Ga,In),Ti2VZ(Z=Al,Ga,In),Ti2YAl (Y=Co,Fe)和Ti2Y’Ga(Y’=Cr,Fe)等合金,都是形成L21结构,而不是形成按照优先占位规则的XA结构[30-32]。
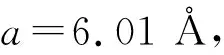
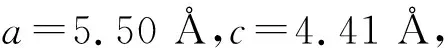
利用第一性原理计算出最稳定的结构为XA立方结构,但实验结果却是DO19六角结构。为此,本文开展了合金的能态密度计算分析。图4为合金总的电子态密度及各原子成分的电子态密度。由图4(a)可见,对于XA相结构,费米能级正好落在自旋向下的态密度的尖峰上,这个尖峰主要来源于Fe,Mn原子3d电子态密度的贡献。通常认为,在接近费米能级处的范霍夫奇点导致态密度高峰,表示结构具有不稳定性[33]。在成分不变的情况下,这种电子结构易引起晶体结构变化,使材料发生相变。如通过晶体结构相变或材料成分微调,来降低费米能级附近的态密度,使费米能级落在某个能谷的地方,体系就达到了一个相对稳定的结构。由图4(b)可见,对于DO19六角结构,费米能级落在2个自旋方向的能谷处,说明这种六角结构可稳定存在。同时,由能量-体积曲线可见,XA相和DO19相之间的能量差只有0.01 eV,理论上来说,只要温度在115 K左右,热扰动就会导致材料变成六角结构,这可能是本文在合成材料的时候,不能出现XA结构的原因,材料只能以DO19六角相结构出现。
另外,在以往的研究中发现,如可能成相的2种结构的能量差较小,则磁性差可能足以克服晶体结构能量的差异,形成能量稍高一点的结构。这就是所谓的磁性会影响晶体结构的形成[34]。通常情况下,磁性交换作用的能量比合金凝固成相能量低约一个量级。如可能成相的2种结构的能量差较小,那么磁性在这种特殊情况下就可能决定成相的种类。在本文合金中,计算得到的能量差仅为0.01 eV,因此,磁性交换作用可能对成相有影响,使材料形成了六角结构。
由此可见,常规的电弧炉熔炼很难合成立方结构。文献曾报道球磨的方法可合成立方结构,且球磨过程中会引入无序和晶格畸变[35]。在之前的研究中发现,晶格畸变会影响费米能级的位置及系统的总能量[36]。如畸变之后系统刚好处在一个能量较低的稳定状态,就会形成立方结构。
温度为5 K时,六角结构Fe2.1MnSn0.9合金的磁滞回线如图5所示,其中,1 Oe=79.5775 A·m-1。由图5可见,合金完全达到了饱和,且矫顽力较小,饱和磁矩约为5.67μB,小于理论计算预测值。根据理论计算,Fe-Mn之间形成了强的铁磁耦合,FeI,FeII和Mn的磁矩分别为2.25μB,2.20μB和2.86μB。在实际样品中,原子之间容易形成反占位,如Fe-Mn,导致部分Mn-Mn反铁磁耦合的出现,因此,饱和磁化强度的测量值比理论值偏低。在Fe2MnGe中也观察到了类似现象[25]。
3 结论
本文通过第一性原理计算分析了Fe2MnSn合金的成相和磁性。理论预测合金以XA型立方结构成相,但XA结构和DO19结构之间的能量差只有0.01 eV,实验发现合金形成DO19型六角结构。进一步的电子结构计算表明,XA相结构的费米能级正好处在态密度的尖峰上,属于不稳定的结构,而DO19结构的费米能级落在态密度的能谷处,是一种稳定的结构。另外,由于二者之间的能量差极小,很小的热能就足以让合金跨过立方结构形成稳定的六角结构,磁性交换作用能也可能影响到晶体结构的形成。这很可能是实验中获得DO19六角相结构的原因。实验获得的分子磁矩小于理论预测值,这主要是因为实验中样品存在反占位,出现了部分反铁磁耦合。
