基于线粒体cytb和d-loop基因序列的东南沿海可口革囊星虫遗传多样性分析
2022-08-11许瑞雯陈兴汉余祥勇
许瑞雯,陈兴汉,余祥勇
(1.阳江职业技术学院,广东阳江 529566;2.广东海洋大学,广东湛江 524088;3.华南农业大学,广州 510642)
可口革囊星虫(Phascolosoma esculenta)为星虫动物门(Sipunculoidea)革囊星虫目(Phascolosomaliformes)革囊星虫科(Phascolosomatidae)革囊星虫属动物,俗称“泥虫”、“土笋”或“泥丁”,是中国特有的星虫种类,在我国东南各省区的沿海滩涂均有分布[1]。可口革囊星虫作为海洋珍品之一,富含蛋白质和多种不饱和脂肪酸等营养物质,被称为“动物人参”,是当地居民的特色经济水产生物。近年来我国东南沿海开发建设进程加快,环境污染、滩涂围垦、超负荷采捕等现象屡禁不绝,导致中国的可口革囊星虫野生种群种质资源量锐减[2],市场价格连年攀升。因此相关的资源保护、种苗繁育及增养殖开发引起行业关注[2],相关学科学者对可口革囊星虫的人工养殖和苗种繁育做了大量研究[3-6]。然而关于可口革囊星虫种群遗传学的研究报道相对较少见,为了该物种资源的合理开发及可持续利用,对其野生群体的遗传多样性开展研究十分必要。
自20世纪60年代线粒体DNA(mtDNA)被发现后[7-8],mtDNA作为核外遗传物质与核基因组DNA相比,由于其具有母性遗传、无重组、分子结构简单、进化速率快、无组织特异性和提取方便等优点[9],已成为研究近缘种和种内群体间遗传分化的有力工具[10-12]。其中,线粒体细胞色素b(cytb)和线粒体控制区(d-loop)两个基因序列是分子标记技术中较常使用的两个基因序列,其遗传多样性在双齿围沙蚕(Perinereis aibuhitensis)[13]、裸体方格星虫(Sipunculus nudus)[14-15]、日本囊对虾(Penaeus japonicus)[16]、厚壳贻贝(Mytilus coruscus)[17]等海洋无脊椎动物的研究中已被应用。
本研究以线粒体DNA的cytb和d-loop基因序列作为分子标记,对我国东南沿海5个可口革囊星虫野生地理群体开展遗传多样性和遗传结构分析,以期为这一中国特有的星虫种类提供人工繁殖和育苗的基础资料,并为后续研究保护该物种的种质资源及其合理开发利用提供数据。
1 材料与方法
1.1 样品信息
实验所用的可口革囊星虫样品均采集于2019年,样品采集地点分别为广东湛江(ZJ)、广东阳江(YJ)、广西钦州(QZ)、福建宁德(ND)、浙江温州(WZ)5个地方的潮间带滩涂,具体采集信息见表1。可口革囊星虫样品均为野生自然个体,系根据该物种的主要形态学特征“大部收吻肌融合,背腹两对相距较远,每对收吻肌的起点较远;虫体颜色为淡黄色或棕色,虫体形状圆长,类似长颈瓶或烧瓶[1]”进行样品鉴定。所用样品均在采样地进行预处理:每个群体采样100个以上,然后每组样品随机选取30个解剖,取其体壁肌肉以95%乙醇固定、保存。
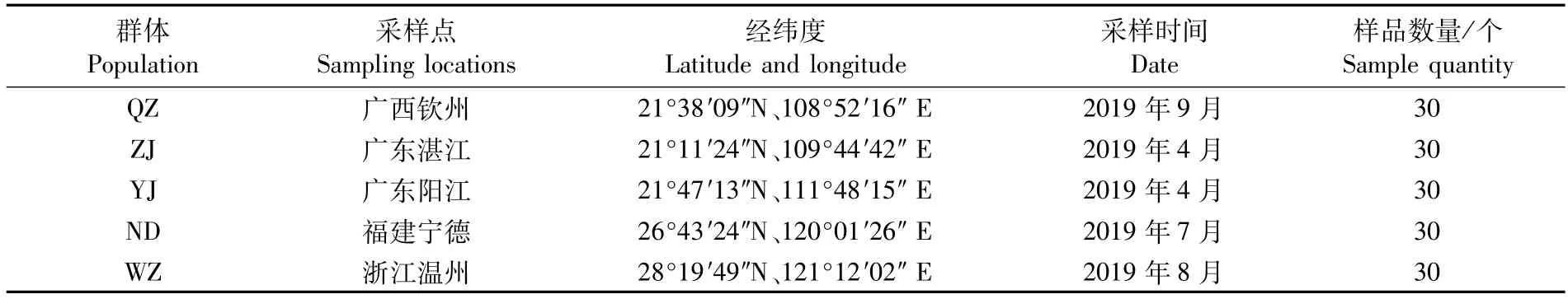
表1 东南沿海可口革囊星虫群体采集信息Tab.1 Information of sampling locations of P.esculenta in southeastern coastal areas
1.2 DNA提取、扩增和测序
取可口革囊星虫体壁肌肉约100 mg剪碎放入1.5 mL的EP(eppendorf)管。采用Universal Genomic DNA Kit(CW2298M,通用型柱式基因组提取试剂盒,康为世纪生物科技股份有限公司)对可口革囊星虫样本进行粗提,然后将提取的DNA样本通过核酸蛋白测定仪(柏点,柏精-BIODROPμLITE,美国)检测,选取DNA浓度大于50 ng·μL-1的样品DNA置于-20℃低温保存,用于后续实验。
根据可口革囊星虫已发布的线粒体基因(GenBank登录号为NC_012618),采用Primer Premier5.0软件[18]进行线粒体cytb序列和d-loop序列的引物设计。其中cytb序列的正、反向扩增引物分别是:PE-F4645(TGGATGACTACTACGATTC)和 PE-R5475(TGGCCTAGTCAGTCATATGG);d-loop序列的正、反向扩增引物分别是:PE-F10225(ACTGAATATTCTAAAGTGGCAG)和PE-R11025(GTGGAAGTCCTATTAGGCTAAC)。以上引物均由广州瑞科基因科技有限公司合成。
根据2×TaqMaster MixPCR扩增试剂盒(code:CW0682L,康为世纪生物科技股份有限公司)的说明书要求和步骤,配置反应体系40μL,其中2×TaqMasterMix(Dye)20μL,正反引物(浓度10μmol·L-1)各2μL,模板DNA 2μL,加灭菌水使反应体系至40μL。PCR反应程序为:94℃预变性3 min;98℃变性15 s,50℃退火30 s,68℃延伸1 min,共35个循环;最后68℃延伸5 min。PCR扩增产物经1%琼脂糖凝胶电泳检测,选出显示亮带的可口革囊星虫cytb和d-loop扩增片段进行纯化测序。
1.3 实验数据分析
测序后的序列使用DNAStar软件包中的EditSeq程序读取,以SeqMan程序对序列进行拼接、编辑,然后用MegAlign程序对序列进行比对并辅以人工校正。修正后的cytb和d-loop序列采用DnaSP 6.0软件[19]对5个地理群体的可口革囊星虫样本分别进行4种碱基构成及数量、多态位点、单倍体数量等运算分析,并检测单倍型在各群体中的分布。
利用MEGA 7.0软件[20]基于K2-P双参数模型计算可口革囊星虫5个群体两两之间的群体间遗传距离,并构建单倍型邻接树(1 000次自展重复)。
利用Arlequin 3.5.2.2软件[21]计算5个群体的基因多样性指数(H)与核苷酸多样性指数(π),并进行分子生物学方差分析(AMOVA分析),估算种群内和种群间遗传变异的分布情况;利用Tajima’s D和Fu’s Fs检验来分析可口革囊星虫群体的历史动态。
2 结果与分析
2.1 可口革囊星虫群体的序列差异及碱基组成
可口革囊星虫的cytb和d-loop基因序列通过PCR扩增,并去除低质量碱基及非目标基因片段后,用于测序的cytb序列碱基长度主要为576 bp,而d-loop序列碱基长度主要为385 bp和384 bp两种,其中大部分是碱基序列,长度为385 bp,占序列总数的75.70% 。通过对序列进行抽样分析可知,序列碱基长度存在多态性的原因是空白位点的插入。经统计,4种碱基含量在可口革囊星虫5个地理群体的cytb和d-loop序列中差异小:cytb的A含量为28.11%~28.17%,G含量为12.70%~12.73%,C含量为25.53%~25.61%,T含量为33.50%~33.62%;d-loop的A含量为38.22%~39.30%,G含量为11.44%~11.58%,C含量为18.76%~18.87%,T含量为31.37%~31.45%。其中两个基因序列均表现出明显的A+T含量高于C+G含量,有明显的碱基偏倚性,与可口革囊星虫的线粒体碱基特征相符[22]。
2.2 可口革囊星虫群体cytb和d-loop序列结构及差异
我国东南沿海5个可口革囊星虫地理群体150个样本个体中:cytb基因序列的多态位点118个,其中转换位点31个,颠换位点87个(颠换和转换比2.81)(表2);d-loop基因序列的多态位点153个,其中转换位点66个,颠换位点87个(颠换和转换比1.32)(表3)。在5个可口革囊星虫地理群体中:广东阳江群体的cytb序列的多态位点最多(31个),福建宁德群体的多态位点最少(16个);广东湛江群体的d-loop序列的多态位点最多(36个),福建宁德群体的多态位点最少(22个)。

表2 可口革囊星虫群体的cytb序列差异Tab.2 Base variation of cytb sequence of P.esculenta populations

表3 可口革囊星虫群体的d-loop序列差异Tab.3 Base variation of d-loop sequence of P.esculenta populations
2.3 可口革囊星虫群体cytb和d-loop序列单倍型组成及分布
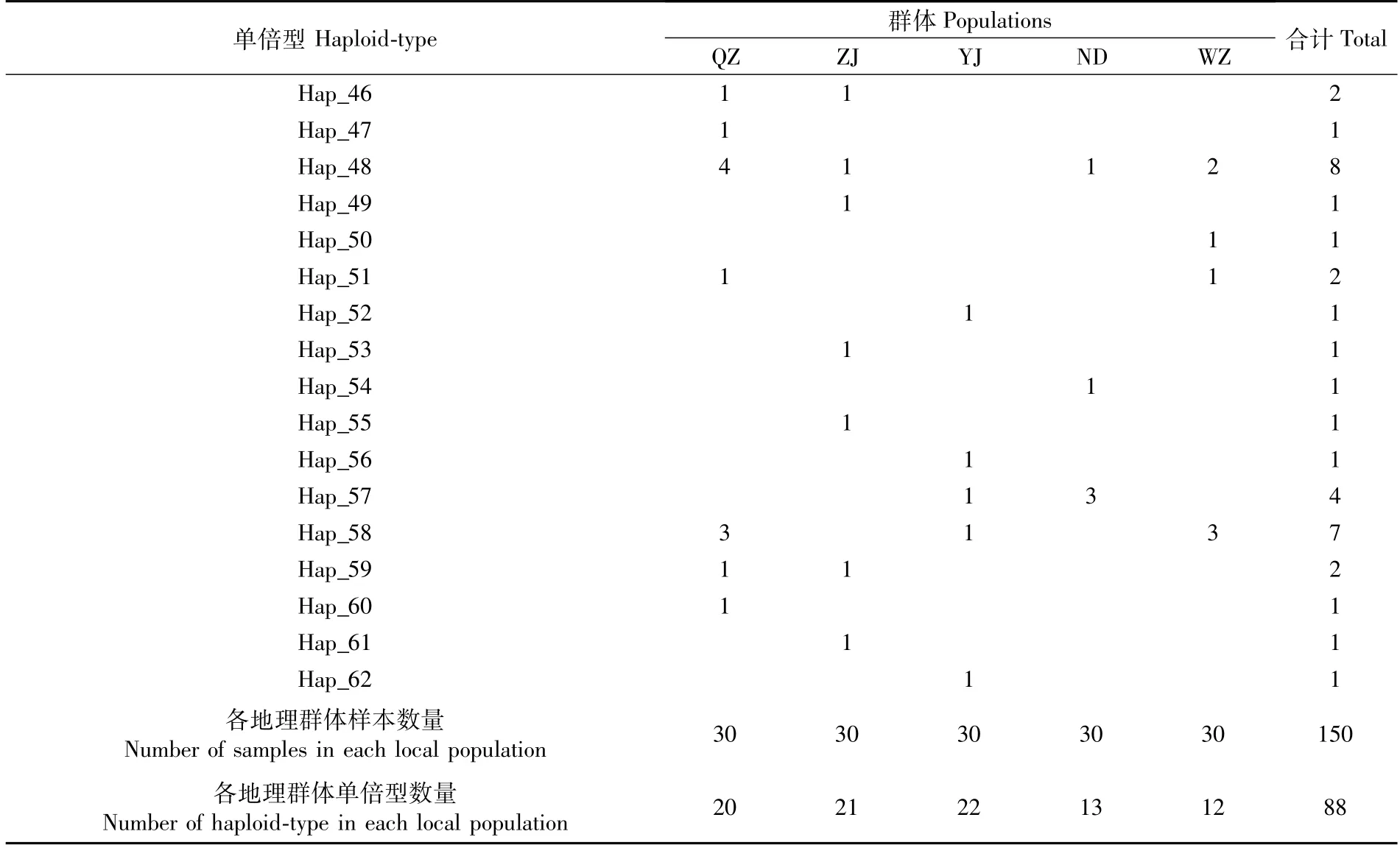
两个基因序列定义的单倍型在各个地理群体间的分布如表4和表5所示。我国东南沿海5个可口革囊星虫地理群体150个样本个体通过cytb序列定义了62种单倍型,通过d-loop序列定义了79种单倍型。
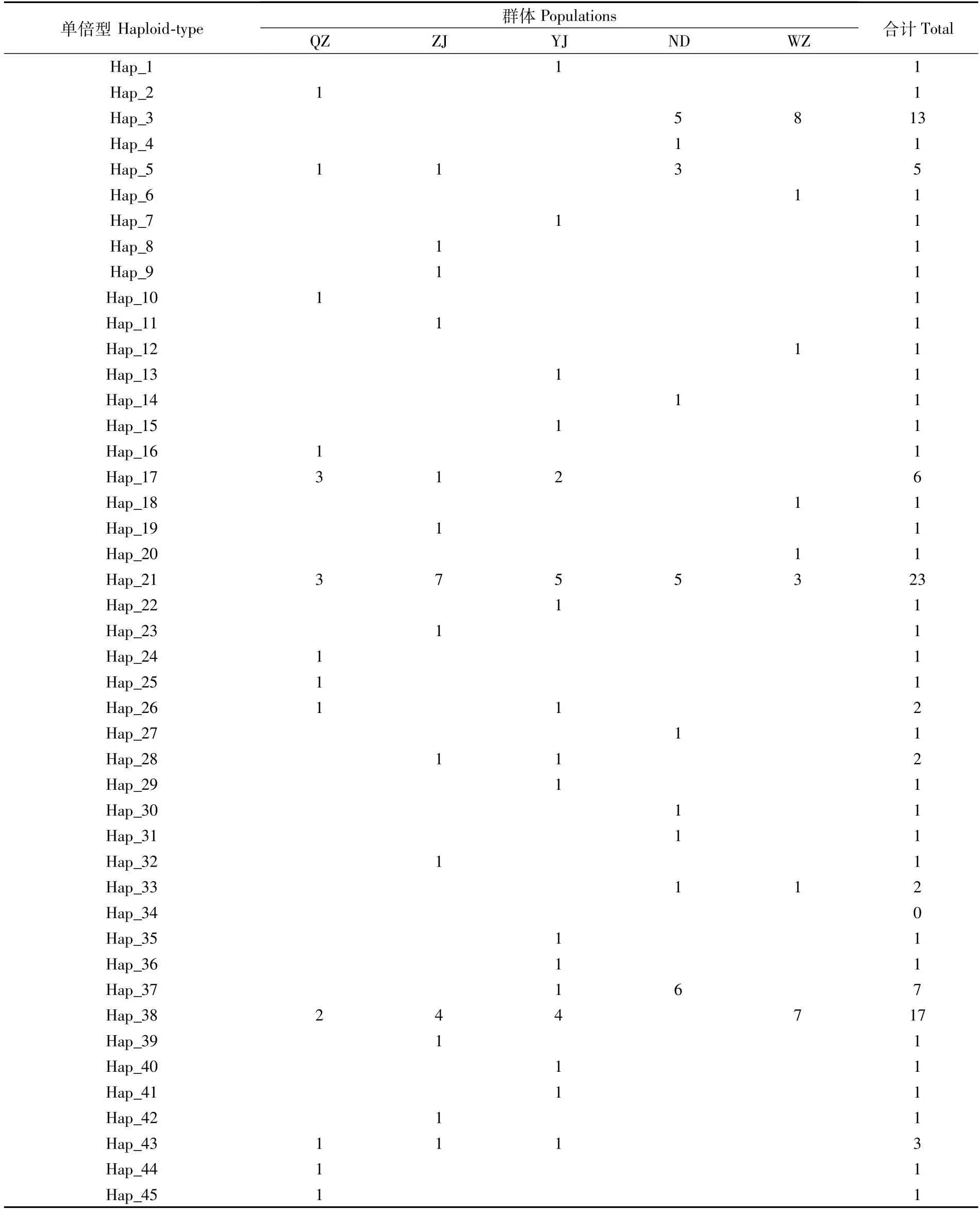
表4 可口革囊星虫群体cytb序列单倍型分布Tab.4 Haplotype distribution of cytb sequence of P.esculenta populations
续表4
通过表4中5个地理群体的cytb序列单倍型数量来看,阳江群体(22个)>湛江群体(21个)>钦州群体(20个)>宁德群体(13个)>温州群体量(12个)。从单倍型的分布情况来看,Hap21在5个地理群体均有分布,也是在5个地理群体中分布最广的单倍型,样本数为23,占总样本数15.33%,其余单倍体大部分分散分布在各个地理群体中。
通过表5中5个地理群体的d-loop序列单倍型数量来看,湛江群体(25个)>阳江群体(23个)>钦州群体(22个)>温州群体(20个)>宁德群体(16个)。从单倍型的分布情况来看,Hap15和Hap67在5个地理群体均有分布,其中:Hap15单倍型的样本数为11,占总样本数7.33%;Hap67单倍型的样本数为17,占总样本数11.33%。其余单倍体大部分分散分布在各个地理群体中。
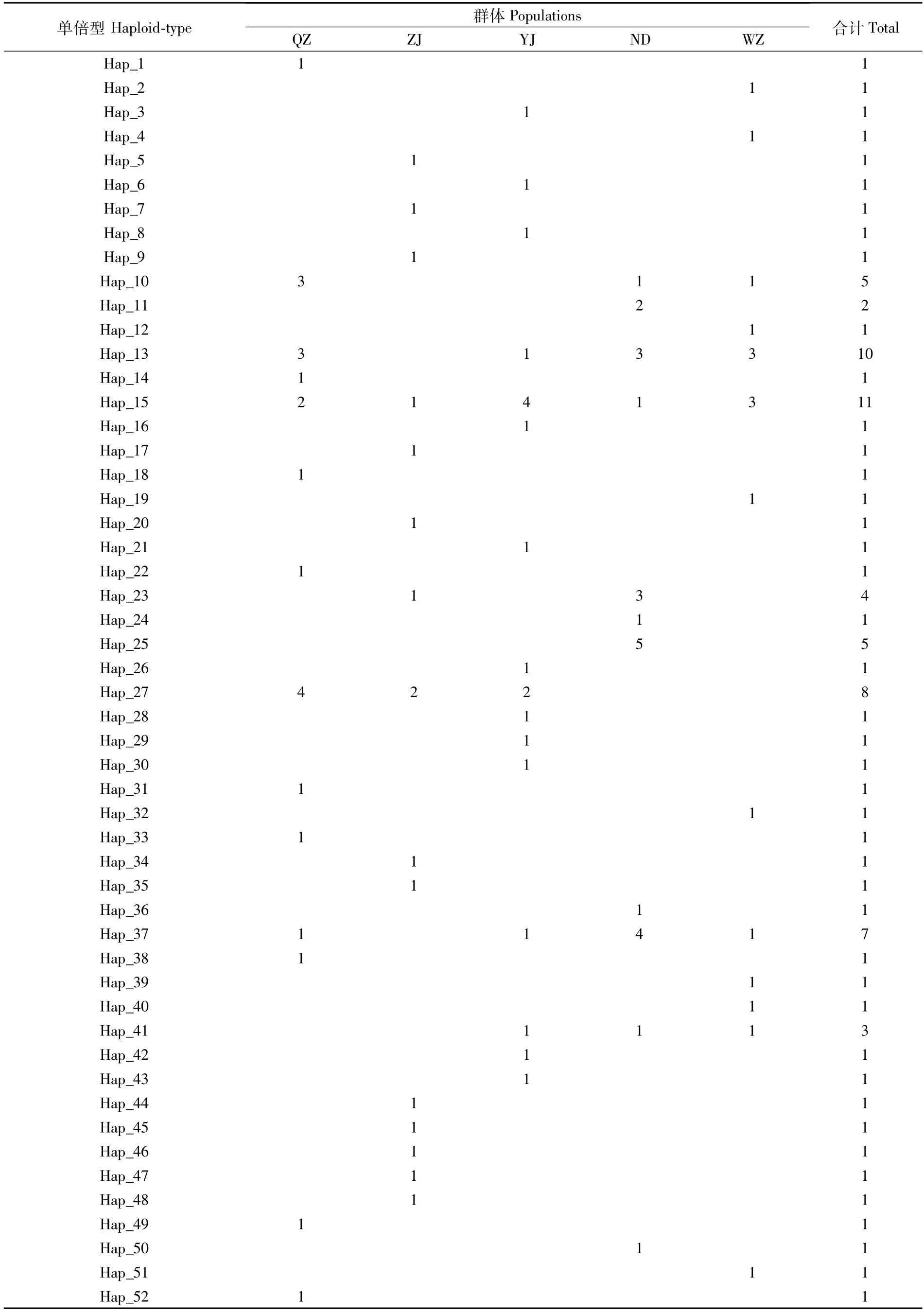
表5 可口革囊星虫群体d-loop序列单倍型分布Tab.5 Haplotype distribution of d-loop sequence of P.esculenta populations
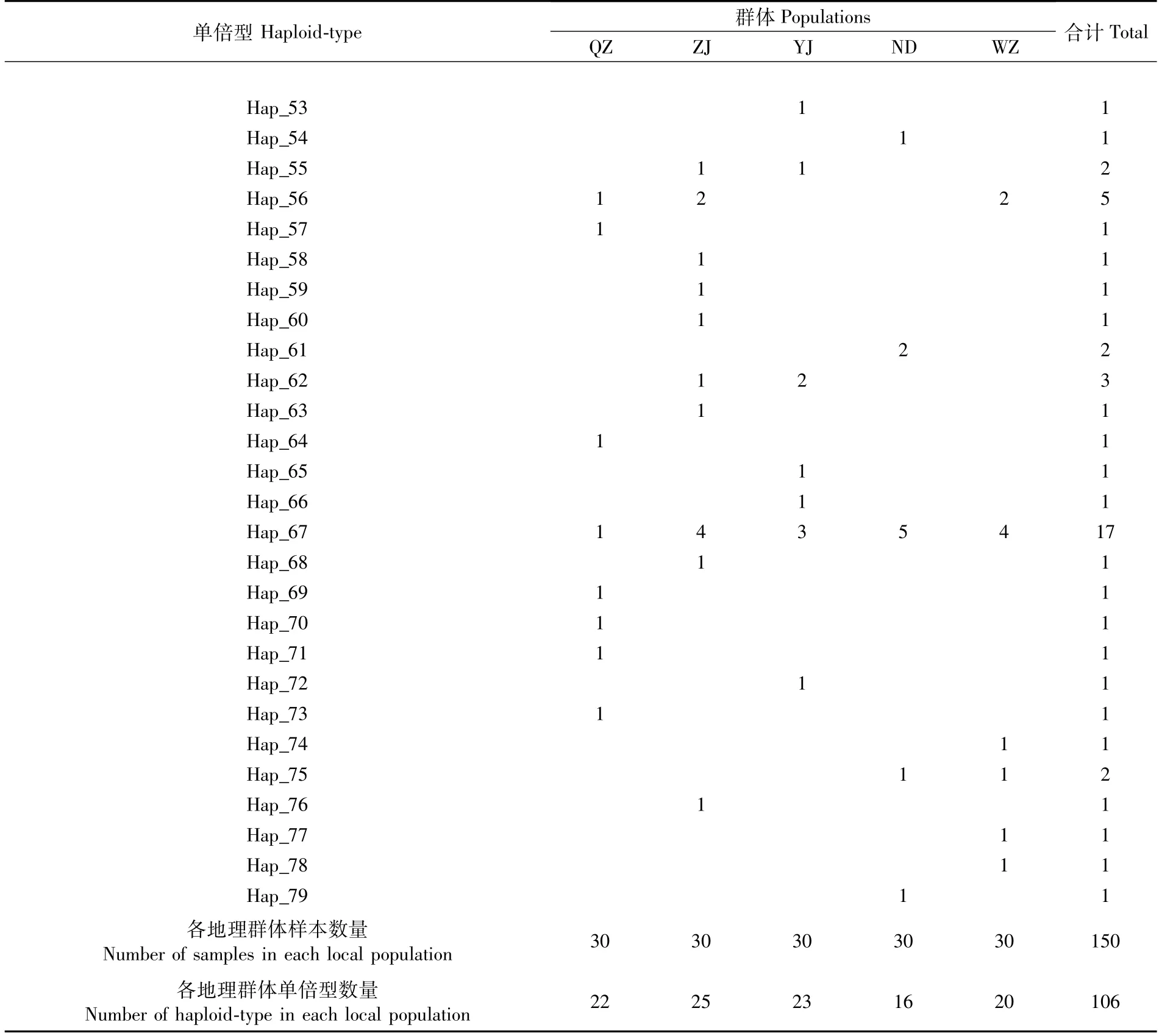
续表5
2.4 可口革囊星虫群体cytb和d-loop序列遗传多样性
如表6所示,可口革囊星虫cytb序列的基因多样性指数(H)为0.871 3~0.963 2,核苷酸多样性指数(π)为0.032 487~0.040 975。其中基因多样性指数(H)和核苷酸多样性指数(π)最高的均为钦州(QZ)群体(H=0.963 2,π=0.040 975)。而基因多样性指数(H)最低的是温州(WZ)群体(H=0.871 3),核苷酸多样性指数(π)最低的则是宁德(ND)群体(π=0.032 487)。

表6 可口革囊星虫cytb序列遗传多样性参数Tab.6 Genetic diversity parameter of cytb of P.esculenta
如表7所示,可口革囊星虫d-loop序列的基因多样性指数(H)为0.942 5~0.981 6,核苷酸多样性指数(π)为0.033 247~0.046 151。其中基因多样性指数(H)和核苷酸多样性指数(π)最低的均是宁德(ND)群体(H=0.942 5,π=0.033 247),而基因多样性指数(H)最高的是湛江(ZJ)群体(H=0.981 6),核苷酸多样性指数(π)最高的则是阳江(YJ)群体(π=0.046 151)。

表7 可口革囊星虫d-loop序列遗传多样性参数Tab.7 Genetic diversity parameter of d-loop of P.esculenta
2.5 可口革囊星虫的群体遗传结构
基于K2-P双参数模型,构建单倍型邻接树(图1,图2)。各地理群体的单倍型均广泛分布在单倍型邻接树上,大部分节点分支支持率较低(<50%),单倍型聚类没有展示出地域性特征。单倍型邻接树的拓扑结构简单,未呈现明显的地理谱系结构。
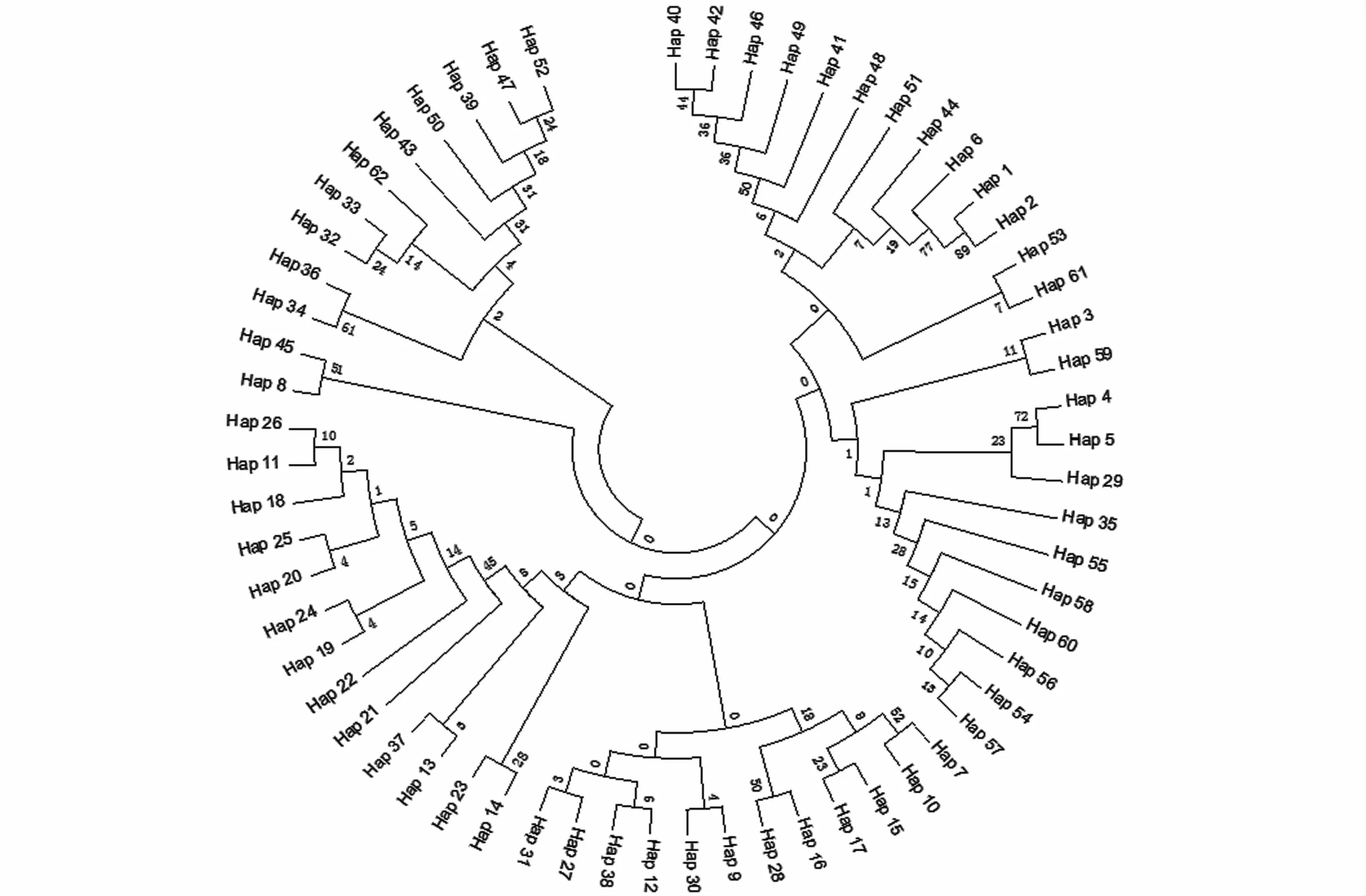
图1 基于可口革囊星虫cytb基因序列构建的单倍型邻接树Fig.1 Neighbor-joining tree of P.esculenta based on cytb gene sequence
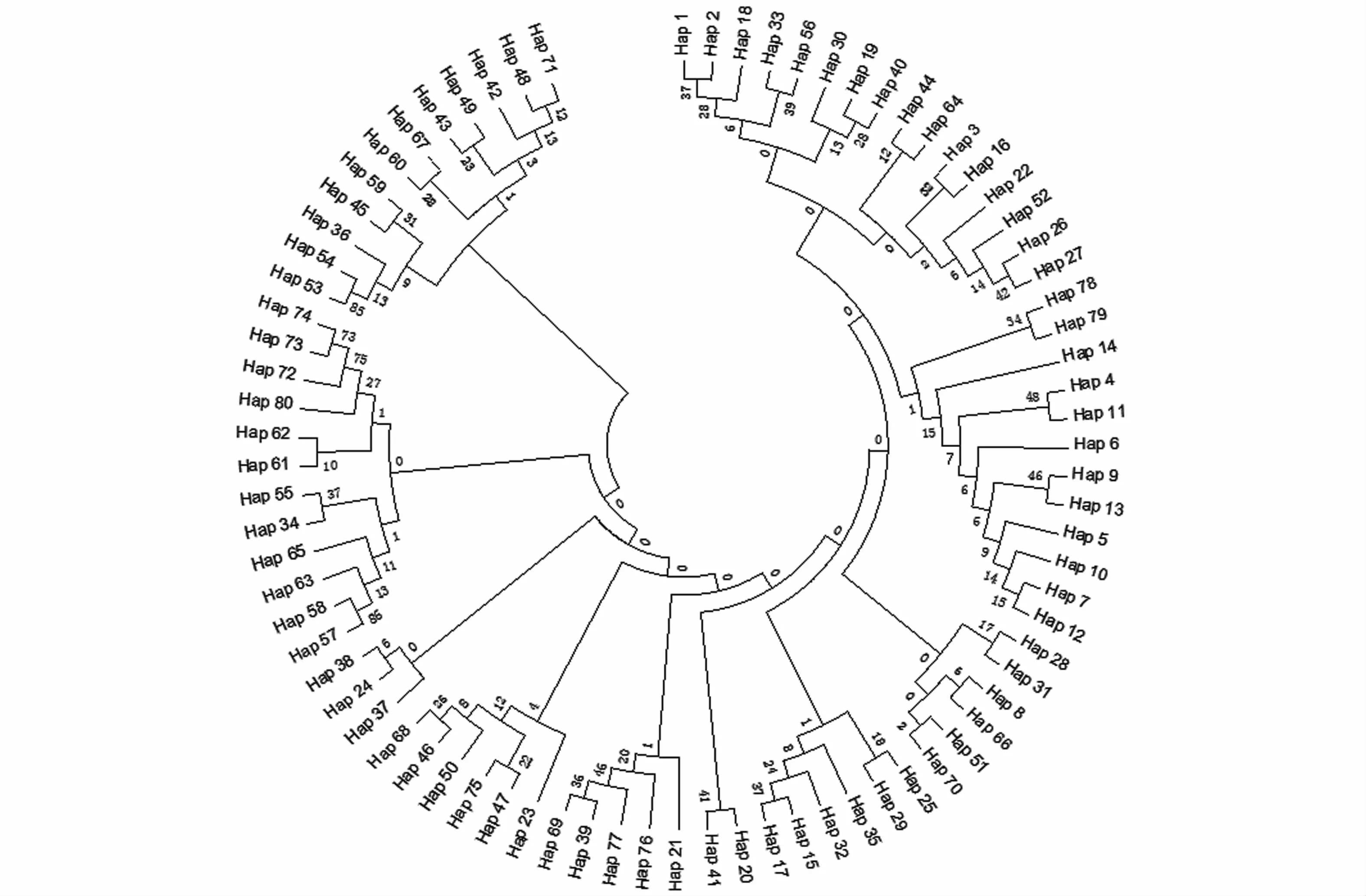
图2 基于可口革囊星虫d-loop基因序列构建的单倍型邻接树Fig.2 Neighbor-joining tree of P.esculenta based on d-loop gene sequence
可口革囊星虫5个群体两两之间的群体间遗传距离如表8所示:基于cytb序列的遗传距离计算结果显示,遗传距离最大的有两对群体(YJ群体和QZ群体,WZ群体和QZ群体)均为0.006 2,最小的也有两对群体(ZJ群体和ND群体,WZ群体和ND群体)均为0.005 4;基于dloop序列的遗传距离计算结果显示遗传距离最大的是YJ群体和QZ群体为0.012 7,最小的是WZ群体和ND群体为0.010 3。
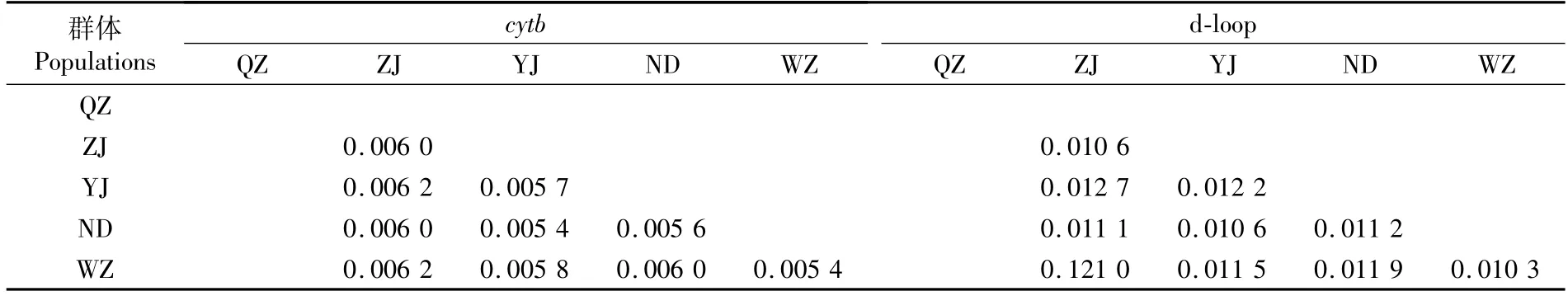
表8 不同群体可口革囊星虫的群体间遗传距离Tab.8 Genetic distance between population of P.esculenta in different populations
如表9所示,不同群体可口革囊星虫cytb和d-loop序列的分子方差分析(AMOVA)结果一致表明,群体内部的遗传变异(cytb:98.56%;d-loop:98.61%)比例显著高于群体间的遗传变异(cytb:1.44% ;d-loop:1.39%)。这表明了可口革囊星虫群体的遗传变异在不同地理群体间并不明显,不存在明显的地理种群分化。

表9 不同群体可口革囊星虫cytb和d-loop序列的分子方差分析Tab.9 AMOVA analysis of cytb and d-loop in different populations of P.esculenta
2.6 可口革囊星虫的群体历史动态
从上文的分子方差分析的结果得出,5个地理群体间的遗传分化不显著。如表10所示,基于cytb和d-loop序列的Tajima’s D和Fu’s Fs检验结果都是负值,并且统计检验均有显著性差异(P<0.05)。综合分析Tajima’s D和Fu’s Fs检验结果显著且偏离中性这一现象,可推测出可口革囊星虫群体近期经历过群体快速扩张事件。
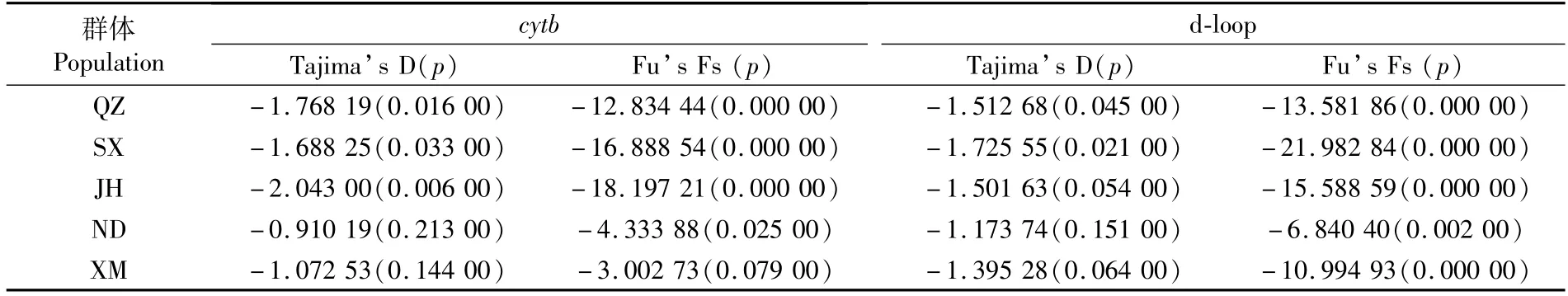
表10 可口革囊星虫cytb和d-loop序列的中性检验Tab.10 Neutral test of cytb and d-loop in P.esculenta
3 讨论
广义定义中的遗传多样性包括了生物所携带遗传信息的总和,不过一般提到的遗传多样性是指单一物种的种内或不同地理群体的遗传变异总和。物种的遗传多样性水平密切影响着该物种适应环境的能力、生存能力和进化潜能等。如多样性水平越高,物种就越能适应栖息地环境的变化。
本次研究通过基因多样性指数(H)和核苷酸多样性指数(π)两个评价遗传多样性的常用参数,对东南沿海的广西钦州(QZ)、广东湛江(ZJ)、广东阳江(YJ)、福建宁德(ND)、浙江温州(WZ)5个地区的可口革囊星虫进行遗传多样性分析。分析结果显示,5个地理群体均表现出基因多样性指数(H)处于较高水平、而核苷酸多样性指数(π)处于较低水平的遗传多样性特征。该特征在金丹璐等[2]基于线粒体COI基因、王帅[23]基于线粒体COI和16SrRNA基因序列研究我国东南沿海其他地理群体的可口革囊星虫的遗传多样性中也有体现:不同地理群体,两个不同序列的分析结果都表现出较高水平的基因多样性和较低水平的核苷酸多样性的分布模式,其原因是基因多样性的累积可在较短时间内提升,但核苷酸变异的累积需要的时间较长[24]。宋素霞等[14]、王剑平等[25]、陈康等[26]研究发现,cytb和d-loop基因序列在双围齿沙蚕(Perinereis aibuhitensis)[13]、光裸方格星虫(Sipunculus nudus)[14]等与可口革囊星虫同为海洋无脊椎类物种的遗传多样性中,都表现出这种高基因多样性水平和低核苷酸多样性水平的特征。该遗传多样性模式在这些海洋无脊柱动物的表现,反映了这些生物群体可能在较近的历史时期经历过快速扩张,即在较短的时间内由一个小规模的繁殖群体衍生为一个大群体[27]。
通过对5个地理群体共150个可口革囊星虫样品的分析,共检测出cytb单倍型类型62种,dloop单倍型类型79种,其中在广东阳江(YJ)和广东湛江(ZJ)两个地理群体中cytb和d-loop两个不同基因的单倍型数量均比另外3个地理群体多,两个不同基因序列的单倍型分布中只有1~2个单倍型在5个群体中均有分布。这表明可口革囊星虫5个地理群体的两种基因序列单倍型随机分散在不同群体中,某些单倍型是多个群体共有,部分单倍型是个别群体特有。这样的单倍型分布现象,从生物学角度可解释为地理群体因一起近期的外来基因入侵事件及随后的遗传分化共同作用产生的结果。
群体遗传结构主要受时间和环境变化的影响,是基因序列的变异在物种或种群中的一种非随机分布,是生物遗传资源应用及物种多样性保护的基础。不同群体可口革囊星虫cytb和d-loop序列的分子方差分析结果以及它们的单倍型邻接树均一致,表明可口革囊星虫群体的遗传变异在不同地理群体间并不明显,说明该物种的5个地理群体没有显著的遗传分化,遗传差异较小,也印证了近年来发表的基于COI基因或cytb基因分析其他地理群体可口革囊星虫遗传变异主要存在于群体间的结论[2,23,28]。导致该现象的原因可能是:1)可口革囊星虫栖息在沿海的潮间带,地理障碍少,而其早期生活史中的担轮幼虫和海球体幼虫时期会随着海水的运动发生远距离扩散,导致不同地理群体之间有了基因交流[29];2)因人工增养殖技术日渐成熟,养殖规模扩大,许多养殖户从多个地方采购苗种投放到当地养殖场,而由于养殖场排水等导致的养殖种群逃逸现象时有发生,使各可口革囊星虫地理群体间的基因交流频繁[2];3)因可口革囊星虫广泛分布在我国东南沿海地区的潮间带,生境条件相似,间接影响了各个不同地理群体的遗传变异方向。另外,通过综合遗传距离的两种算法结果,福建宁德(ND)群体与其他群体两两之间的遗传距离是最近的,可能与当地养殖场人工投放的苗种逃逸导致基因混淆有关[2]。因为采样点牛姆屿所在的内海湾是闽东地区最大的可口革囊星虫养殖基地——霞浦县长春镇土笋养殖基地,因当地采挖的星虫苗种满足不了养殖需求,养殖户从广西、浙江等地大量采购苗种投放养殖,由于潮汐的影响人工苗种逃逸现象必然存在,这导致当地野生地理群体的基因混淆现象严重。
综上分析,广东湛江(ZJ)、广东阳江(YJ)、广西钦州(QZ)、福建宁德(ND)、浙江温州(WZ)5个可口革囊星虫地理群体两个不同序列的分析结果都表现出较高水平的基因多样性和较低水平的核苷酸多样,总体遗传多样性水平较高;遗传变异在不同地理群体间并不明显,说明该物种的5个地理群体没有显著的遗传分化,遗传差异较小;另外,从两个不同序列的Tajima’s D和Fu’s Fs检验结果可推测出,可口革囊星虫群体近期经历过群体快速扩张事件。本研究结果可以为可口革囊星虫种质资源的保护与可持续开发利用提供参考数据。
