液相色谱串联质谱法测定人血浆中硫酸黏菌素的浓度
2022-08-09马颖超董维冲吴瑕杨秀岭河北医科大学第二医院药学部石家庄050000
马颖超,董维冲,吴瑕,杨秀岭(河北医科大学第二医院药学部,石家庄 050000)
黏菌素(又称多黏菌素E)是一种含多种组分的阳离子多肽类抗菌药物,于1950年从大肠埃希菌中分离得到,其结构是脂肪酸链通过酰胺键与阳离子十肽环连接。黏菌素主要成分为多黏菌素E1(C53H100N16O13)和多黏菌素E2(C52H98N16O13),两者区别仅是脂肪酸侧链相差一个甲基,多黏菌素E1为6-甲基辛酸,多黏菌素E2为6-甲基庚酸[1],这两种成分占黏菌素总含量的85%以上[2],决定其主要血药浓度和临床抗菌活性[3]。硫酸黏菌素,原名硫酸抗敌素,又称硫酸多黏菌素E,是1966年我国自主研发的抗菌药物。硫酸黏菌素与多黏菌素E 甲磺酸盐不同,无需转化,直接杀菌。因市场用量少,曾经停产。近年来,由于临床耐药菌的广泛出现,硫酸黏菌素对多重耐药及泛耐药革兰氏阴性杆菌特别是铜绿假单胞菌、鲍曼不动杆菌和肺炎克雷伯菌有强大的抗菌活性,被作为治疗这些多重耐药菌感染的最后一道防线,于2018年重新上市,用于危重患者严重感染的治疗[4]。但由于其上市较早,未经历过现代药物开发程序,早期的药动学、药效学、毒理学等资料非常有限,缺少指导临床使用的相关信息。其治疗窗窄,不良反应发生率高[5-6],尤其在重症感染患者中,临床个体差异大,需要进行血药浓度监测[7],以便实现个体化给药。本文建立一种液相色谱串联质谱(LC-MS/MS)法测定人血浆中硫酸黏菌素主要成分多黏菌素E1和多黏菌素E2的浓度,现报道如下。
1 材料
1.1 仪器
LC-20C 高效液相色谱仪(日本岛津),API4000+三重四级杆质谱仪(Applied Biosystem,美国),离心机(德国ABBOTT),涡旋混合器(XW-80A,上海医科大学仪器厂),低温冰箱(MDF-U2086S,日本三洋),冷藏柜(YC-180,澳柯玛),电子分析天平(CPA225D,德国Sartorius)。
1.2 试药
硫酸黏菌素对照品(含量:95.1%,批号:833621)、内标硫酸多黏菌素B(含量:88.1%,批号:174224)(德国Dr Ehrenstorfer 公司);人血浆(河北省血液中心);甲酸(色谱纯,上海Aladdin);乙腈(色谱纯,美国Fisher 公司);水为蒸馏水。
2 方法与结果
2.1 色谱条件
色谱柱:Dikma C18色谱柱(4.6 mm×150 mm,5 μm)。流动相A:水(含0.1%甲酸),流动相B:乙腈(含0.1%甲酸),梯度洗脱(0 ~1 min,95%A;1~5 min,25%A;5~7 min,5%A;7 ~9 min,95%A);流速:0.8 mL·min-1;柱温:40 ℃;进样量:10 μL。
2.2 质谱条件
电喷雾离子源(ESI),多重反应离子监测(MRM)正离子模式,喷雾电压(IS):5500 V,离子源温度(TEM):550℃,气帘气压力(CUR):10 psi,雾化器压力(GAS1):55 psi,辅助气压力(GAS2):55 psi,碰撞气压力(CAD):4 psi。多黏菌素E1、多黏菌素E2和内标多黏菌素B 的定量离子对分别为m/z585.7 →101.2、m/z578.8 → 101.2 和m/z602.7 → 241.4,去簇电压(DP)分别为61 V、59 V 和68 V,碰撞电压(CE)分别为47 V、49 V 和33 V。
2.3 对照品溶液和内标溶液的配制
精密称取硫酸黏菌素、硫酸多黏菌素B 对照品各20 mg,分别置于10 mL 量瓶中,用20%甲醇溶解并稀释成质量浓度为2 mg·mL-1的硫酸黏菌素储备液和内标储备液。精密量取硫酸黏菌素储备液适量,以20%甲醇逐级稀释成质量浓度为1、2、5、10、20、50、100 μg·mL-1的系列标准工作液和质量浓度为1、10、80 μg·mL-1的质控工作液,用20%甲醇稀释内标储备液为40 μg·mL-1作为内标工作液,4℃冰箱储存备用。
2.4 血浆样品处理
取血浆200 μL,加入20 μL 内标工作液(40 μg·mL-1),涡旋混匀后加入400 μL 甲醇-5%三氯乙酸(50∶50,V/V),涡旋2 min,10 900 r·min-1离心5 min,取上清液200 μL 进样。
2.5 方法学考察
2.5.1 专属性 取人空白血浆、人空白血浆+硫酸黏菌素对照品储备液(5 μg·mL-1)+内标工作液(40 μg·mL-1)、临床患者用药后血浆样品,按“2.4”项下方法处理分析。结果多黏菌素E1、多黏菌素E2和内标的保留时间分别为5.64、5.54和5.68 min。峰形良好,血浆中内源性物质在待测物保留时间处无干扰,结果见图1。
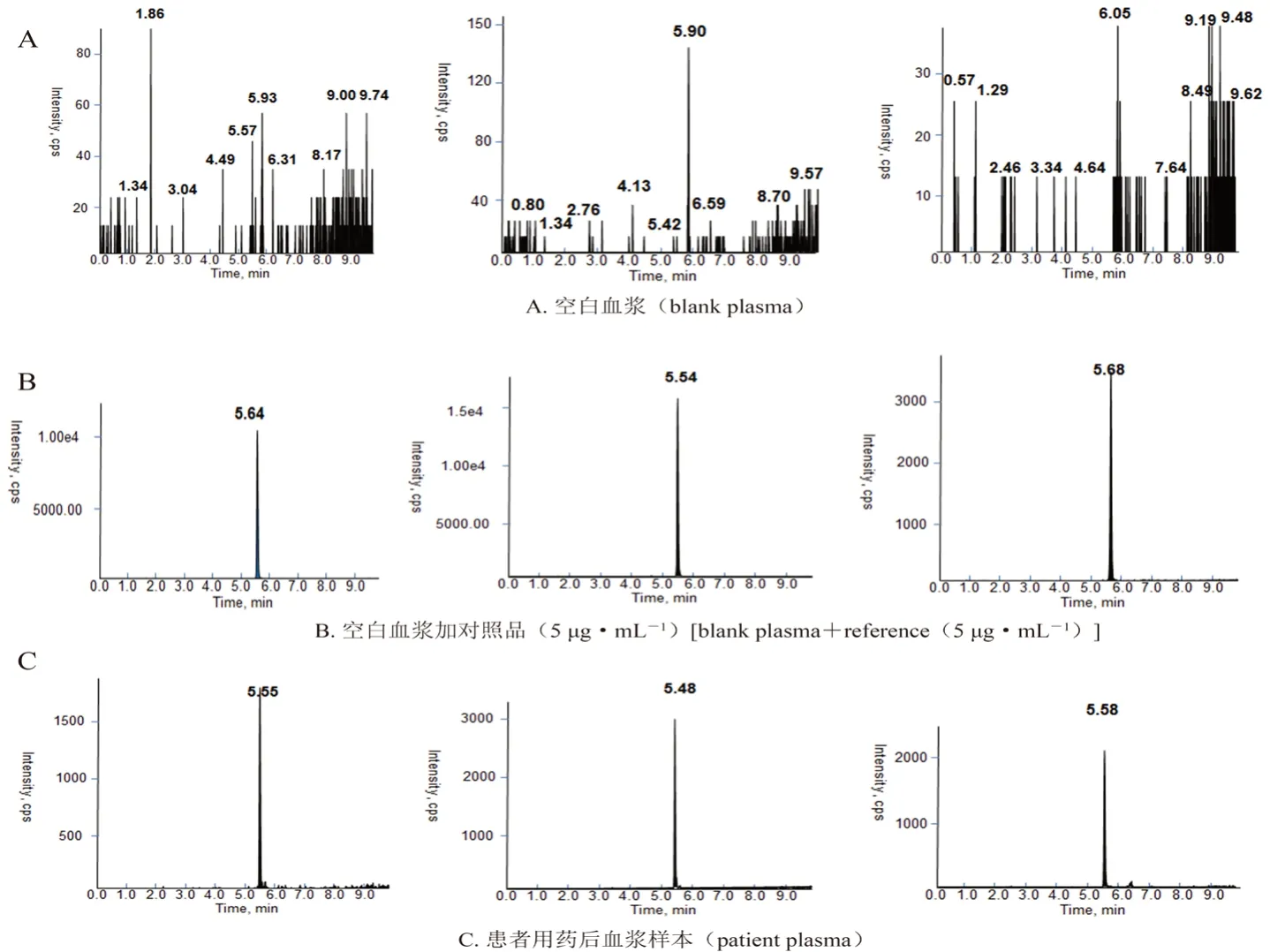
图1 硫酸黏菌素的HPLC-MS/MS 色谱图Fig 1 HPLC-MS/MS chromatogram of colistin sulfate
2.5.2 标准曲线与定量限 将“2.3”项下配制好的硫酸黏菌素系列标准工作液分别加入到空白血浆中制备成质量浓度为0.1、0.2、0.5、1、2、5、10 μg·mL-1的标准曲线溶液。按“2.4”项下方法处理后进样,以多黏菌素E1和多黏菌素E2总浓度为横坐标,多黏菌素E1和多黏菌素E2总峰面积与内标物的峰面积比值为纵坐标,以加权最小二乘法进行线性回归运算,得硫酸黏菌素标准曲线方程为y=1.851x-0.059(r=0.9982),在0.1 ~10 μg·mL-1与峰面积线性关系良好。定量限(LOQ)为0.1 μg·mL-1。
2.5.3 精密度与准确度 取“2.3”项下硫酸黏菌素系列质控工作液,分别加入到空白血浆中制得低(0.1 μg·mL-1)、中(1 μg·mL-1)、高(8 μg·mL-1)质控工作液,按“2.4” 项下方法处理,每个浓度平行做5 份,连续测定3 d,根据当日标准曲线计算日内和日间精密度、准确度,结果见表1。
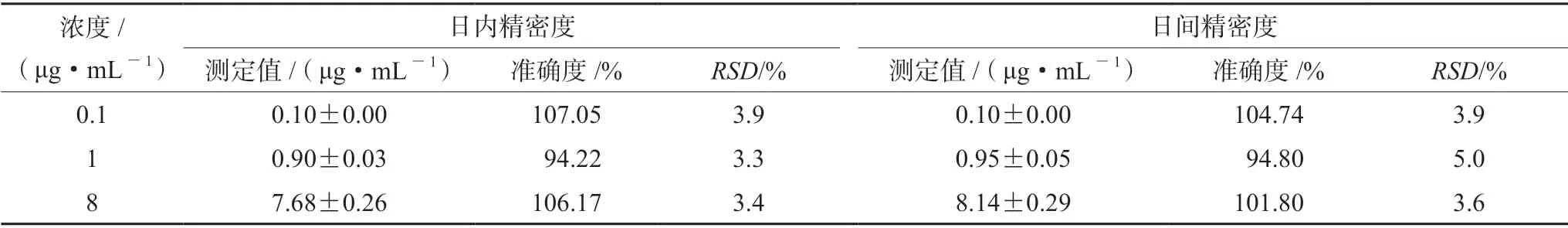
表1 精密度和准确度试验结果(n =5)Tab 1 Precision and accuracy tests (n =5)
2.5.4 提取回收率和基质效应 取“2.5.3”项下3个浓度的质控工作液,按“2.4” 项下方法处理,作为样品A;取空白血浆进行预处理后的上清液,加入系列质控工作液,制得低、中、高质控工作液,作为样品B;另配制同浓度标准溶液,作为样品C,每个质控浓度各5 份。将3 组样品同法进行HPLCMS/MS 分析,用样品A 的峰面积与样品C 的峰面积比计算提取回收率,样品B 的峰面积与样品C的峰面积比计算基质效应,结果见表2。
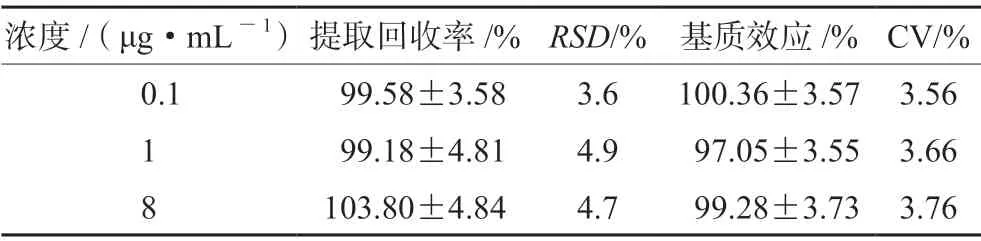
表2 提取回收率和基质效应试验结果(n =5)Tab 2 Extraction recovery and matrix effect (n =5)
2.5.5 稳定性试验 制备低、中、高3 种质量浓度硫酸黏菌素质控血浆样品,考察分别经过室温放置6 h 和24 h,自动进样器15℃放置24 h,-20℃冻融循环3 次,-70℃冰箱放置30 d,按“2.4” 项下方法处理后测定血浆样品的稳定性,得到硫酸黏菌素浓度的RSD均在±15%,稳定性符合相关要求,结果见表3。
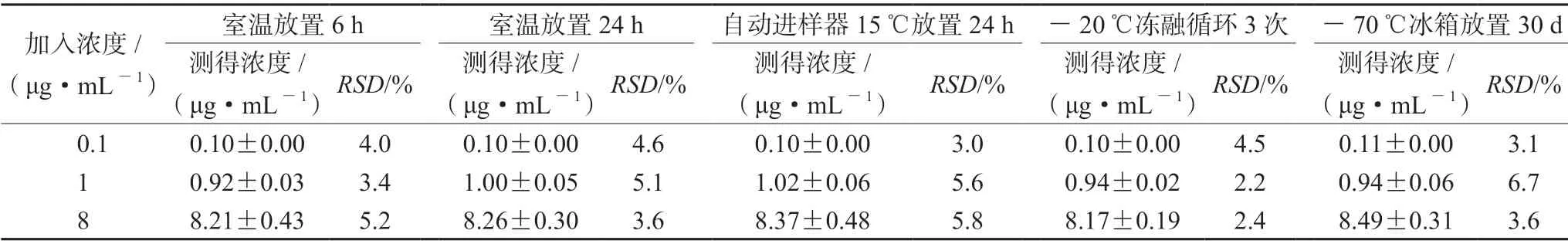
表3 稳定性试验结果(n =5)Tab 3 Stability test (n =5)
2.6 方法学验证
用本研究所建方法检测10 例患者硫酸黏菌素血药谷浓度,平均年龄为(53.20±24.36)岁,给药剂量按照说明书规定静脉滴注每次50 万IU,每12 h 给药一次,其中3 例患者给予首剂加倍。硫酸黏菌素谷浓度应在稳态下进行测定,其半衰期在肾功能正常的患者中为6 h,根据药代动力学理论,需经过4 ~5 个半衰期达到稳态浓度,故可在用药2 ~3 d 达稳态后取血测定,最终选定第7 剂给药前30 min 采集血样,测定硫酸黏菌素稳态谷浓度,结果中最大浓度为1.44 μg·mL-1,最小浓度为0.25 μg·mL-1。同时抽取了其中两例患者第7 剂给药后15 ~30 min 血样,测定硫酸黏菌素峰浓度,分别为1.96 μg·mL-1和1.79 μg·mL-1,结果见表4。
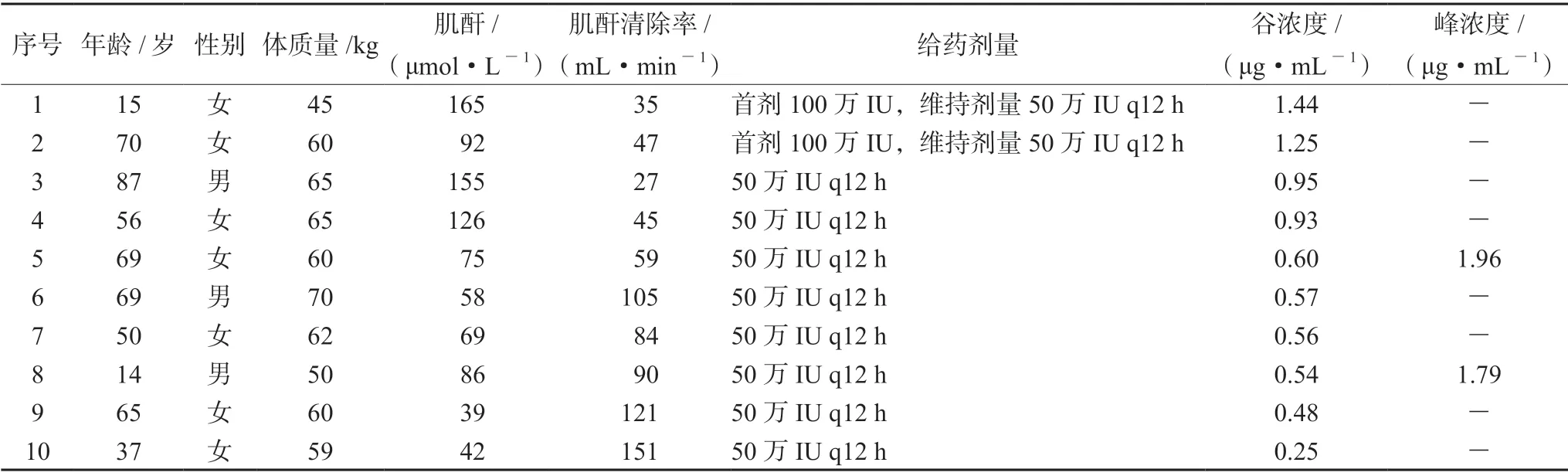
表4 10 例患者硫酸黏菌素峰谷浓度测定结果Tab 4 Cmax and Cmin of colistin sulfate in 10 patients
3 讨论
3.1 硫酸黏菌素临床应用困惑
硫酸黏菌素曾因其肾毒性和神经毒性遭到临床弃用,仅应用于兽药中。但随着多重耐药菌感染在临床逐渐增多,在新药开发有限的前提下,硫酸黏菌素又被重新应用于临床[8]。但由于硫酸黏菌素未经历过现代药物开发程序,缺乏合理应用所需要的药理学信息,目前对各类患者特别是重症患者,其给药方案并没有得到循证医学的证实[9];并且目前应用较少,经验不足,临床医师对硫酸黏菌素的使用存在很大困惑。一方面很多临床医师不完全清楚硫酸黏菌素的给药剂量,原因是各药企所生产的多黏菌素E 有着不同的标签和不同的给药方案,有些为国际单位,有些标有“多黏菌素E 基础活性”的标签。这种在标签以及每日推荐剂量上的不一致,造成临床医师对多黏菌素E 的两种常用剂型黏菌素甲磺酸盐和硫酸黏菌素的使用剂量产生混乱,对硫酸黏菌素的临床使用有着非常大的影响。另一方面,近年来很多研究指出硫酸黏菌素的肾毒性发生率和严重程度远低于早期临床观察结果,仅在联用其他肾毒性药物或剂量过高时可增加肾功能损害发生的可能性[10-12]。文献报道,硫酸黏菌素导致的肾功能损害是可逆的,停药后数周至数月内可恢复[13]。尽管如此,在使用硫酸黏菌素时仍需密切监测肾功能且避免联用有肾毒性的药物。药动学和药效学分析可明晰药物在体内的吸收、分布、代谢和排泄,从而指导临床用药剂量和给药方式,降低不良反应发生率,因此,当硫酸黏菌素被重新应用于临床后,对其进行药动学和药效学研究显得越发重要。
3.2 硫酸黏菌素血药浓度分析方法选择
现有的多黏菌素E 血药浓度分析方法有微生物法、免疫法、薄层色谱法、毛细管电泳法[14]和高效液相色谱法(HPLC)[15]。微生物法样本耗时多且缺乏特异性,免疫法免疫原制备复杂,分析成本高,薄层色谱法和毛细管电泳法重复性差,不具有普遍性[1]。由于多黏菌素E 最大吸收波长为210 nm,有末端吸收,此波长处流动相中的有机相会有一定吸收,因此对检测器灵敏度和流动相要求苛刻。紫外检测器会造成基线漂移,数据不可信。荧光检测器虽然专属性和灵敏度高,但由于多黏菌素E 没有原生荧光,需要进行提取和衍生化,操作繁琐[16]。本文采用LC-MS/MS 法测定人血浆中硫酸黏菌素的浓度,灵敏度高,准确度好,符合《中国药典》生物样本检测方法的要求。
文献[14-16]中对多黏菌素E 样品前处理大多采用固相萃取法,但该技术操作繁琐,不利于大量生物样品的检测,且固相萃取板价格昂贵。本研究采用蛋白沉淀样品的前处理方式,简化了血浆预处理过程,节约了成本,适用于临床大量样本分析。在方法学优化过程中考察了多种流动相,包括水-甲醇、水-乙腈、水(含0.01%甲酸)-乙腈(含0.01%甲酸)、水(含0.1%甲酸)-乙腈(含0.1%甲酸)、水(含0.5%甲酸)-乙腈(含0.5%甲酸)、水(含1%乙酸)-乙腈(含1%乙酸),最终选择本文流动相。通过加入0.1%甲酸使得多黏菌素E1和多黏菌素E2色谱峰峰形良好,响应值得到改善。在洗脱方法研究中,选择梯度洗脱,首先采用较低比例的有机相冲洗,让极性物质先流出色谱柱,保证了极性物质和药物先后流出,避免了基质效应的干扰,并通过调整初始有机相比例得到了较为合适的保留时间和峰形,提高了检测灵敏度。在内标选择中,选择与黏菌素结构和化学性质相似的多黏菌素B,保证完全分离的同时,又可以获得相似的回收率、离子化效率和色谱保留行为,并且无内源性物质干扰和基质效应干扰。
3.3 硫酸黏菌素血药浓度检测结果分析
多黏菌素E 血药浓度的高低与抗菌效果、肾毒性紧密相关,重症患者体内多黏菌素E 药动学变化大,血药浓度监测不仅能保证治疗效果,也能降低肾脏损害的风险。2019年多黏菌素国际共识中建议[17],多黏菌素E 的推荐靶值为稳态时24 h 血浆药时曲线下面积(AUCss,24h)达到50 mg·h·L-1,即稳态平均血药浓度(Css,avg)维持在2 μg·mL-1。该浓度被认为是能耐受的最高暴露量,高于此浓度将增加急性肾损伤的发生率和严重性。通过本研究方法检测10 例患者硫酸黏菌素血药谷浓度在0.25 ~1.44 μg·mL-1,谷浓度差异较大。其中5 号和8 号患者平均稳态血药浓度低于指南要求,患者硫酸黏菌素给药剂量为50 万IU q12h,符合说明书规定,且患者肾功能正常,但平均稳态血药浓度不能达标,推测给药剂量可能偏低,但由于本研究峰浓度样本量较少,该结论尚需更多峰谷浓度测定来证实。另外,1 号和2 号患者采用首剂加倍给药方案,其谷浓度分别为1.44 μg·mL-1和1.25 μg·mL-1,明显高于未采用首剂加倍的患者。计算患者肌酐清除率,1、2 号患者均存在肾功能不全,硫酸黏菌素主要经肾排泄,用药后8 h 内尿中排出用药量的40%,肾功能不全者,药物易在体内蓄积,故血药谷浓度升高是首剂加倍和肾功能不全两者同时导致的。但与同样肾功能不全、未采用首剂加倍的3、4、5 号患者相比,谷浓度也明显升高,说明首剂加倍可提高硫酸黏菌素的血药浓度。
综上,硫酸黏菌素体内个体差异较大,患者肾功能情况可影响药物排泄,说明书规定50 万IU q12h 的给药方案可能存在剂量不足,需采用首剂加倍的给药方案来增加血药浓度,提高临床疗效。但由于本研究纳入病例数和取血点有限,其他影响硫酸黏菌素体内药动学的因素尚需更大样本量、更全面的临床研究证实,本研究所建方法为进一步研究硫酸黏菌素药动学-药效学性质提供了参考。
