微波消解电感耦合等离子体质谱法对地表水中痕量磷的监测研究
2022-08-08何云勇豆艳霞马贻翠
何云勇,豆艳霞,马贻翠
(安康市环境保护监测站 陕西 安康 725000)
磷(P)是所有生物生长和能量传递所必需的营养元素,通常是陆地和水生生态系统初级生产者的限制性营养元素[1-3]。自然界存在丰富的磷源,其主要储存在陆地环境,水环境和大气环境相对较少,但在各种生态系统中随能量和物质交换不断地迁移转化,在生物地球化学循环中发挥着重要的作用。在过去的几十年里,由于人类文明发展过程中对磷的过度使用而导致的富营养化和水华已经成为一个世界性的环境问题[4]。各种人类活动,包括农业、废水处理、城市扩张和工业发展,是磷过度使用的主要因素。
磷在自然水体中有多种存在形式,有研究从实验操作上根据是否能够通过0.45 μm/0.2 μm滤膜,将其区分为总溶解态磷和总颗粒态磷[5]。根据磷的化学形态,总溶解态磷和总颗粒态磷均可分为无机磷和有机磷两种。
磷在水生生态系统中与多种无机/有机物质结合,以多种形式存在,生物活性差异显著。磷酸盐是水环境中最稳定、最丰富的磷形态,可被细菌、藻类、浮游植物等直接利用,是生物最容易获得的磷形式,也是首要可被吸收的磷源[6]。由于不同的浮游植物对磷酸盐的需求不同且相互竞争,水体中磷酸盐浓度的变化可能导致浮游植物群落在季节或水华过程中发生更替[7]。
总磷(TP)是指水样中存在的所有含磷化合物含量的总和,包括正磷酸盐、磷单酯(C-O-P 键)、磷二酯(C-P 键)、膦酸酯(C-P 键)、聚磷单酯(C-OP-O-P-O-P 键)、聚磷酸盐(P-O-P-O-P 键)、焦磷酸盐(P-O-P 键)[8]。总磷代表了生物可利用磷的最大潜在量[9]。因此,在许多标准中总磷是一个重要的环境指标因子,可以作为水质指标和废水处理过程中的控制参数。
我国现行《地表水环境质量标准》(GB 3838—2002)中规定地表水中总磷的标准限值,见表1。总磷的分析方法采用《水质总磷的测定钼酸铵分光光度法》(GB 11893—1989)(以下简称国标法),方法检出限为0.01 mg/L。由于标准限值与要求的检出限接近,在水质评价时存在争议。有研究指出,湖库地表水中总磷量超过0.02 mg/L 就会造成富营养化,美国环保局认为中营养化阶段总磷为0.01~ 0.02 mg/L,达到0.02~ 0.03 mg/L呈富营养化状态[10]。
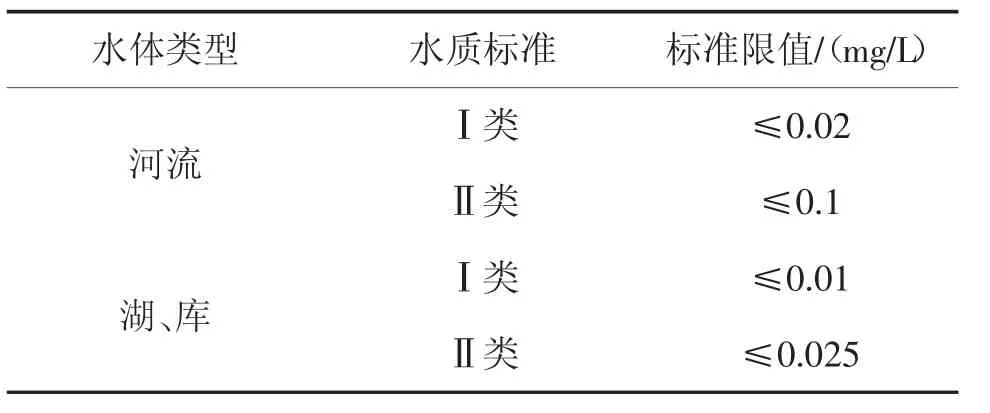
表1 地表水中总磷的标准限值Tab.1 Standard limits for total phosphorus in surface water
随着环境保护政策的进一步加强,管理要求的不断提高,监测数据需更加精准,同时现行标准分析方法检出限为0.01 mg/L,对于全国水体环境总磷监测要求太高,针对I 类地表水总磷监测过程中“检出即超标”,特别是对湖库标准无法适应问题,急需开发新的磷测定分析方法。本文主要探讨溶解性磷和总磷的监测分析方法。
1 磷监测过程探讨
根据监测目的,水环境中磷的监测选择不同的方法。水体中磷/总磷检测原理不同,可以将其分为分光光度法[11]、离子色谱法[10]、电感耦合等离子体法[12]。不同的分析方法都有学者做过一定实验研究,各自有优缺点。受监测过程中样品采集保存和前处理的方式影响,不同方法的检出限、测定范围也不同,因此在探讨水环境样品中磷/总磷测定时不能忽略样品采集保存和前处理过程。
1.1 样品采集
在采样之前,重要的是要确保采样策略适合于监测的目的[13]。包括确定适当的采样地点(适当考虑到道路和安全问题)、采样频率以及监测哪种形态的磷。这就需要明确说明采样方案的目标,了解磷的生物地球化学和磷形态[14]的稳定性。
采集样品可以手工采集(离散采单点样品或断面样品),也可以通过自动采样器进行时间序列采集。同时在每个地点/时间段采集平行样。有研究提出在单点样品至少应采集分析3 个子样品,以保证定量测定稳健性分析[15]。
采样点布设考虑磷浓度的时间和空间变化,并确保所采集的样本具有被采样水体的代表性。水体磷浓度空间变异受点源及面源扩散输入、水中过程(如植物、藻类和细菌周转)、混合区、湖泊中随深度的热分层或河口中的盐度分层的影响。时间变异受到河流流量和物理化学梯度(例如温度和盐度)的季节性(如夏季径流量大于冬季)和短期(例如降雨事件)变化的影响[16]。
清洁的样品容器和采样器是减少污染的必要先决条件。采样器具的清洁方法均涉及酸清洗,如在无磷洗涤剂中浸泡一夜,用超纯水漂洗,然后在10%的盐酸中浸泡一夜,最后用超纯水[16]冲洗。样品容器首选聚四氟乙烯(PTFE)或高密度聚乙烯(HDPE)材料。考虑价格因素,一般也可选用石英、硼硅酸盐玻璃、低密度聚乙烯和聚丙烯等材质。采样的最小体积一般根据分析方法确定,建议至少500 mL。有研究者建议使用更大尺寸和更小表面积与体积比[17]的容器可以使吸附产生的损失最小化。灌装样品时,应将样品容器使用样品荡洗3 次后,装满整个容器。在采集样品时,最好使用样品空白来监测取样过程。
1.2 保存与前处理过程
地表水测定总磷时加硫酸或者硝酸至pH<1,测定溶解性正磷酸盐时不加任何保存剂,样品于4 ℃以下保存,尽快分析。
测定总磷时水样的处理方式为消解,一般为消解试剂+加热的组合方法。常用的消解试剂为过硫酸钾/过硫酸钠、硝酸/高氯酸。加热方式有微波加热[10]、高压蒸汽锅加热[11]、水浴加热及烘箱加热[18]、紫外加热等[19]。另外也有研究者提出高温燃烧消解法,即在550 ℃下将所有有机物转化成无机盐类[20],溶解后进行测试。
不同的消解方法对不同形态的磷转化成正磷酸盐的效率不同。过硫酸钾/过硫酸钠+高压蒸汽消解测定总磷,通过选取6 种含磷有机物模拟消解过程,发现不同的消解试剂对不同化合物消解效果不同。存在一些含磷化合物在两种试剂及两种加热作用下均不能完全消解。这说明在传统的消解方式下,仍然有部分磷未转换成正磷酸盐,这将会导致后续测试时测定的正磷酸盐并非全部的总磷。Zhang[21]指出过硫酸盐消解法对不同的溶解性磷化合物的转换效率不同,例如肌醇磷酸盐转化成正磷酸盐只有66%,相对湿法消解更推荐高温燃烧法的干法消解方式。
1.3 测试方法
分光光度法的基本依据是朗伯比尔定律,总磷国标法的显色原理是在酸性环境中,正磷酸盐与钼酸铵在锑盐存在下发生化学反应生成磷钼杂多酸,再加入抗坏血酸为还原剂,将其还原成蓝色络合物。此方法已有很多研究报道[22-24],通过改变溶液pH、更换显色试剂、更换还原剂等方式以提高方法灵敏度,通过增加比色皿厚度、萃取显色物等方法提高准确度。然而方法的弊端依然存在,在实际样品测定中色度、浊度的干扰依然是影响实验结果的不可避免的因素。通常采用补偿法、过滤法和离心法去除干扰。但对于低浓度样品,如果以色度-浊度补偿试验会产生较大干扰,数据精密度、准确度无法保证。
离子色谱法的基本原理是将水体中各种形式的磷转化成正磷酸盐,正磷酸盐在色谱柱中随强碱性淋洗液进入阴离子色谱柱,以磷酸根的形式被分离出来后,用电导检测器检测[25]。根据保留时间定性,外标法定量。这种方法的前提仍然是需要前处理步骤,而且在消解后高浓度的硫酸盐妨碍磷酸盐的测定。这可能是以前许多使用离子色谱研究磷酸盐测定时,避免使用碱性过硫酸盐消解或开发出一种更复杂的方法以最小化掩蔽这种干扰的最常见原因[26]。朱红霞等[10]将环境监测实验室常用的微波消解前处理技术和离子色谱测定方法相结合,优化微波消解和离子色谱条件,方法检出限为0.002 mg/L。
电感耦合等离子体发射光谱法主要过程为:经过滤或消解的水样注入电感耦合等离子体发射光谱仪后,目标元素在等离子体火炬中被气化、电离、激发并辐射出特征谱线,在一定浓度范围内,其特征谱线的强度与元素的浓度成正比[12]。该方法测定磷的研究也有诸多报道。方法线性范围宽,可以和其他元素同时测定;检出限较高,适合废水中总磷的测定。
电感耦合等离子体质谱法也是一种测定水体中各种元素的方法,其原理是水样经预处理后,采用电感耦合等离子体质谱进行检测,根据元素的质谱图或特征离子进行定性,内标法定量[27]。待测元素随进样系统进入等离子体炬管后,再在高温电离环境下转化成带电荷的正离子,经离子采集系统进入质谱仪,质谱检测器根据离子的质荷比进行分离并定性、定量。该方法应用总磷检测的研究较少,主要受限于仪器发展的水平,以及对仪器操作人员有一定要求。
2 ICP-MS 测定磷的实验研究
2.1 主要仪器试剂
仪器:ICAP-RQ 型ICP-MS,美国赛默飞世尔科技公司;723 型分光光度计,上海菁华科技有限公司;LDZF-50KB-Ⅱ型立式压力蒸汽灭菌器,上海申安医疗器械厂;MARS6 微波消解仪,美国培安科技有限公司(CEM)。
试剂:硝酸(优级纯,经2 次亚沸提纯,科密欧试剂公司);过硫酸钾(优级纯,SIGMA 公司);高氯酸(优级纯,国药集团化学试剂有限公司);磷标准储备液(国家有色金属及电子材料分析测试中心)。
2.2 样品采集
样品采集于湖库、河流原有固定监测断面,用于测试的样品分组采集:SA1#~SA10#加1%硝酸保护剂,SB1#~SB10#不加保护剂,SC1#~SC10#以0.45 μm 滤膜现场过滤后加1%硝酸保护剂。所有样品盛装在经硝酸浸泡24 h 并清洗干净的聚四氟乙烯采样瓶中。所有组别均采集全程序空白水样。
2.3 样品处理
所有样品按照2 种处理方式。过硫酸钾消解:取水样25 mL 置于比色管中,加入饱和过硫酸钾4 mL,塞盖以纱布包紧后置于高压蒸汽锅内(1.1 kg/cm2,121 ℃)保持30 min。微波消解:20 mL 水样加入到聚四氟乙烯消解管,加入高氯酸1 mL,设置消解温度为120 ℃,消解时间实验优化后确定。
3 实验结果讨论
3.1 方法参数验证
ICP-MS 测定磷,提前调整预热仪器,仪器参数按照仪器自动调谐的结果使用,元素选择31P,配置浓度为1.0 μg/L、5.0 μg/L、10.0 μg/L、50.0 μg/L、100.0 μg/L、500.0 μg/L 的曲线浓度点,保持2%硝酸酸度。内标选择45Sc,在线添加,内标浓度为20 μg/L。线性回归的结果为:Y=8.444X+9.515,相关系数r=0.9999。曲线最低点计算浓度与实际浓度相对偏差为6.5%。
分别以低浓度实际水样平行测试7 次,计算标准偏差δ,以3 倍的标准偏差δ 认定为方法的检出限,即0.4 μg/L;4 倍检出限认定为测定下限,即1.6 μg/L。
正确度验证,以国家环境保护部标准样品研究所标样验证,结果均在保证值范围内。平行样验证相对偏差为6.5%,实际水样加标浓度为5 μg/L,加标回收率为93.2%。
以上结果说明ICP-MS 可以用于水中痕量磷的测定。
3.2 前处理优化
国标法中应用过硫酸钾作氧化剂,高压蒸汽锅消解法已经应用多年,存在高温高压消解安全隐患、消解时间长等缺点。本实验研究微波消解,提高效率,降低风险。
实验分别采用标准样品和实际样品在微波消解管中消解,评定在不同消解时间下的测定结果。分别采用ICP-MS 和光度法测定消解完成的水样。总磷质控样品编号为200540,浓度为(193±22) ug/L,经稀释后进行消解测定,结果见表2。

表2 不同消解时间下测定结果Tab.2 Determination results under different digestion time
从表2 可以看出,质控样无论消解时间多久,ICP-MS 法测定结果总能在给定范围内,用光度法测定时消解10 min 以上,结果能够在给定范围内。以实际样品消解测定,15 min 能完全消解。因此,为提高样品分析的效率,建议水样消解时间为15 min。
3.3 不同监测方法对结果的影响
根据不同的采样预处理、消解方法、分析方法,分析地表水断面样品,结果见表3。
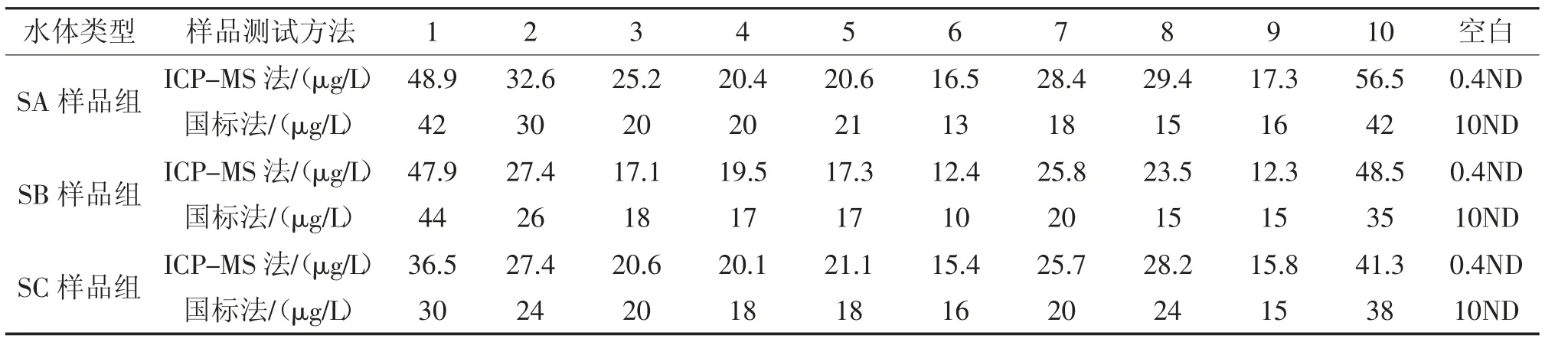
表3 不同采样点位监测结果Tab.3 Monitoring results of different sampling points
从表3 可知,未经过滤测定水样(SA 组)样品磷的测定结果比经0.45 μm 过滤处理的水样(SC组)磷含量高,说明水体中悬浮颗粒物吸附有部分含磷物质,水样过滤会使部分水样磷含量监测结果偏低,这符合实际情况。
微波消解ICP-MS 测定磷的结果,总体比国标光度法测定的结果高,这可能与消解方式和测定方式有关。表2 数据也表明测定质控样品时,微波ICP-MS 法对消解时间没有限制,测定值均能在给定范围内;光度法必须消解完全后,测定值才能在给定范围内。采用过硫酸钾+高温蒸汽消解的方式测定结果总体偏小,可能是由于水样中磷未完全转化成正磷酸所致。而使用ICP-MS测定时,可以不考虑消解不完全的问题,只要样品能够进入ICP 等离子体中,在高达几千摄氏度的温度下,所有含P 化合物电离成P+,进而均能检测到。同时对于较清洁地表水可以不用前处理,直接进样检测。
加酸保存的水样(SA 组)比没有加酸保存的水样(SC 组)监测出磷的浓度高,说明水样采集和保存方法对监测结果有重要影响;样品采集后磷会有一定的变化,因此加酸保护是重要的步骤。数据表明,样品采集后加酸保护能够促进磷在水体中的稳定,防止吸附在容器表面导致监测结果偏小。
4 结论与展望
水体中磷的监测要根据监测目的,考虑采样过程中的预处理和保存剂的使用。总磷监测过程中加1%的硝酸保存是可行的方法。本文比较了过硫酸钾+高温蒸汽消解+分光光度法与微波消解ICP-MS 法测定水中的磷。应用微波消解后ICP-MS 法测定水中微量磷,曲线范围为1.0~500 μg/L,线性相关系数为0.9999,检出限为0.4 μg/L,能够满足Ⅰ类、Ⅱ类水体(特别是湖、库)低浓度磷的监测。同时优化了微波消解时间,提高分析效率。实验发现在一些监测过程中使用滤膜过滤水样会使水样监测浓度偏低,使用国标分析方法也会低估了水体总磷的含量。微波消解ICP-MS法基于检测过程中高温电离环境能够克服样品前处理消解不完全的问题,同时可以不考虑水样色度带来的影响,是一种值得推广的方法。
随着仪器水平不断进步,本论文中还有许多地方需要进一步研究,如:水样的盐度、有机物含量对仪器的干扰、ICP-MS 本身多原子离子干扰消除、消解后酸度控制对仪器灵敏度的影响等方面。
