加味香连丸HPLC指纹图谱和11个成分定量分析
2022-08-08钟国男张余芳钟小丽
钟国男,张余芳,林 冠,钟小丽
(海口市妇幼保健院 药剂科,海南 海口 570203)
加味香连丸为清热剂,是由木香、黄连(姜炙)、黄芩、黄柏(酒炙)、白芍、当归、厚朴(姜炙)、枳壳(去瓤麸炒)、槟榔、延胡索(醋炙)、吴茱萸(甘草炙)、甘草(蜜炙) 十二味中药组成[1]。方中黄连,清热燥湿,止泻痢,为君药;黄芩、黄柏加强黄连清热燥湿之功,共为臣药;白芍、当归和血止痛,延胡索理气止痛,厚朴、枳壳、槟榔、木香行气和中,行滞止痛,吴茱萸温中燥湿止泻,为佐药;甘草健脾和中,调和药性,为使药;诸药合用,共奏清热祛湿,化滞止痛之功。临床用于抗生素相关性腹泻[2]、放射性肠炎[3]、溃疡性结肠炎[4]。加味香连丸收载于2020年版《中华人民共和国药典》(以下简称《中国药典》),现行标准仅对盐酸小檗碱含量做限量要求,对木香、厚朴、枳壳和延胡索进行薄层定性鉴别。有关其质量控制的文献主要集中于芍药苷、柚皮苷、和厚朴酚、厚朴酚、黄芩苷、木香烃内酯和去氢木香内酯等成分的测定[5-7]。
中药制剂为多组分复杂体系,现行质量标准中薄层鉴别及含量测定均不能对加味香连丸进行整体描述和评价。中药指纹图谱是中药或中药制剂经适当处理后,借助一定的分析手段,得到的能标示其化学特征的色谱图或光谱图,能较为全面地反映中药及其制剂中所含化学成分的种类与数量,进而对药品质量进行整体描述和评价,具有“整体性”和“模糊性”的特点,已广泛应用于中药材和中药制剂的质量控制[8-11]。目前加味香连丸指纹图谱研究尚未报道。本研究采用高效液相色谱(HPLC)法建立加味香连丸的指纹图谱,并对其中的芍药苷、柚皮苷、新橙皮苷、阿魏酸、木香烃内酯、去氢木香内酯、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素11个主要活性成分的含量进行分析,为全面控制加味香连丸的质量提供实验依据。
1 仪器与材料
1.1 仪器
Agilent 1260高效液相色谱仪(美国Agilent公司);BSA124S万分之一分析天平(北京赛多利斯科学仪器有限公司);XS205十万分之一电子天平(瑞士梅特勒-托利多公司);KQ-250DBG型数控超声波清洗器(功率:250 W,频率:40 kHz,昆山超声波仪器有限公司)。
1.2 药品与试剂
芍药苷对照品(批号:110736-201842,含量以97.4 %计),柚皮苷对照品(批号:110722-201815,含量以91.7 %计),新橙皮苷对照品(批号:111857-201804,含量以99.4 %计),阿魏酸对照品(批号:110773-201614,含量以99.0 %计),木香烃内酯对照品(批号:111524-201911,含量以99.9 %计),去氢木香内酯对照品(批号:111525-201912,含量以99.5 %计),黄芩苷对照品(批号:110715-201720,含量以93.5 %计),盐酸小檗碱对照品(批号:110713-202015,含量以85.9 %计),和厚朴酚对照品(批号:110730-201915,含量以99.8 %计),厚朴酚对照品(批号:110729-202015,含量以99.0 %计),延胡索乙素对照品(批号:110726-201819,含量以99.8 %计),均购自中国食品药品检定研究院。加味香连丸[北京同仁堂股份有限公司同仁堂制药厂,批号分别为2080882(S1),20810197(S2),3080036(S3),3081265(S4),3082242(S5),3086576(S6),3086783(S7),4082251(S8),4083197(S9),4084062(S10),6080152(S11),6082278(S12),6087253(S13),60897421(S14),8080524(S15),规格为每100丸重6 g。甲醇、乙腈为色谱纯,磷酸为分析纯,实验用水为娃哈哈纯净水。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent SB-C18(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.05 %磷酸水溶液(B),梯度洗脱(0~5 min,15 %A;5~20 min,15 %A→35 %A;20~55 min,35 %A→60 %A;55~65 min,60 %A;65~75 min,60 %A→80 %A;75~80 min,80 %A→5 %A),流速为1.0 ml/min,检测波长230 nm(8~12 min)、225 nm(29~32 min)、280 nm(0~8 min,12~29 min和32~80 min),柱温30 ℃,进样量10 μl,色谱记录时间80 min。
2.2 混合对照品溶液的制备
分别精密称取芍药苷、柚皮苷、新橙皮苷、阿魏酸、木香烃内酯、去氢木香内酯、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素对照品,置量瓶中,分别加入甲醇溶液制备成质量浓度分别为1.374,7.915,2.339,1.537,0.888,0.847,11.947,17.584,1.085,3.019,0.355 mg/ml的单一对照品储备液。分别精密量取上述单一对照品储备液适量,置同一10 ml量瓶中,加入70 %甲醇溶液稀释并定容至刻度,摇匀,经0.45 μm的微孔滤膜滤过,即得浓度分别为137.4,791.5,233.9,153.7,88.8,84.7,1194.7,1758.4,108.5,301.9,35.5 μg/ml的混合对照品溶液,备用。
2.3 供试品溶液的制备
取加味香连丸供试品适量,研细,取约1.0 g,精密称定,置100 ml具塞锥形瓶中,精密加入70 %甲醇30 ml,称定重量,超声处理(功率250 W,频率40 kHz)30 min,放冷,再次称定重量,用70 %甲醇补足减失的重量,摇匀,经0.45 μm微孔滤膜滤过,取续滤液即为供试品溶液,备用[12]。
2.4 指纹图谱方法学考察
2.4.1 精密度试验 取加味香连丸供试品(批号:2080882),按2.3项下方法制备供试品溶液,按2.1项下色谱条件连续进样6针,记录色谱图及峰面积,以13号色谱峰黄芩苷的保留时间为参照,计算得各共有峰的相对保留时间RSD均小于1.3 %(n=6),相对峰面积RSD均小于1.9 %(n=6),说明仪器精密度良好。
2.4.2 重复性试验 取同一批加味香连丸供试品(批号:2080882)6份,每份约1 g,按2.3项下方法制备供试品溶液,按2.1项下色谱条件进样分析,记录色谱图及峰面积,以13号色谱峰黄芩苷的保留时间为参照,计算得各共有峰的相对保留时间RSD均小于1.1 %(n=6),相对峰面积RSD均小于1.5 %(n=6),说明本方法重复性良好。
2.4.3 稳定性试验 取加味香连丸供试品(批号:2080882),按2.3项下方法制备供试品溶液,分别于室温下放置0,4,8,12,16,24 h,按2.1项下色谱条件进样分析,记录色谱图及峰面积,以13号色谱峰黄芩苷的保留时间为参照,计算得各共有峰的相对保留时间RSD均小于1.5 %(n=6),相对峰面积RSD均小于1.6 %(n=6),表明供试品溶液在室温条件下24 h内稳定。
2.4.4 指纹图谱的建立及相似度评价 取15批加味香连丸供试品,按2.3项下方法分别制备成供试品溶液,按2.1项下色谱条件进样分析,记录HPLC色谱图。采用《中药色谱指纹图谱相似度评价系统》(2012A版)(以下简称《相似度评价系统》)软件评价分析,以编号S1号样品的色谱图为参考图谱,设置时间窗宽度0.2 min,采用多点校正全谱匹配法建立指纹图谱共有模式,并以平均值法生成对照图谱。结果共标定29个共有峰,15批加味香连丸指纹图谱与对照指纹图谱的相似度分别为0.997,0.993,0.992,0.998,0.996,0.991,0.999,0.995,0.995,0.996,0.994,0.995,0.999,0.997,0.992,相似度均大于0.99,说明15批加味香连丸化学成分一致性较好。指纹图谱及其共有模式见图1。

图1 15批加味香连丸HPLC指纹图谱
指纹图谱共获得29个保留时间较稳定的共有峰,通过比对混合对照品溶液色谱图共指认出11个化合物,分别是3号色谱峰芍药苷、4号色谱峰柚皮苷、5号色谱峰新橙皮苷、8号色谱峰阿魏酸、11号色谱峰木香烃内酯、12号色谱峰去氢木香内酯、13号色谱峰黄芩苷、21号色谱峰盐酸小檗碱、26号色谱峰和厚朴酚、27号色谱峰厚朴酚、28号色谱峰延胡索乙素,其中13号色谱峰保留时间相对居中,与相邻色谱峰分离度好,峰高和峰面积较适中,因此选择13号峰(黄芩苷)作为参照峰,计算其他共有峰的相对峰面积和相对保留时间[13-14]。结果29个共有峰的相对保留时间RSD均小于1.90 %(n=15),相对峰面积RSD为0.90 %~21.27 %,具体数据见表1。对照品溶液色谱图见图2。
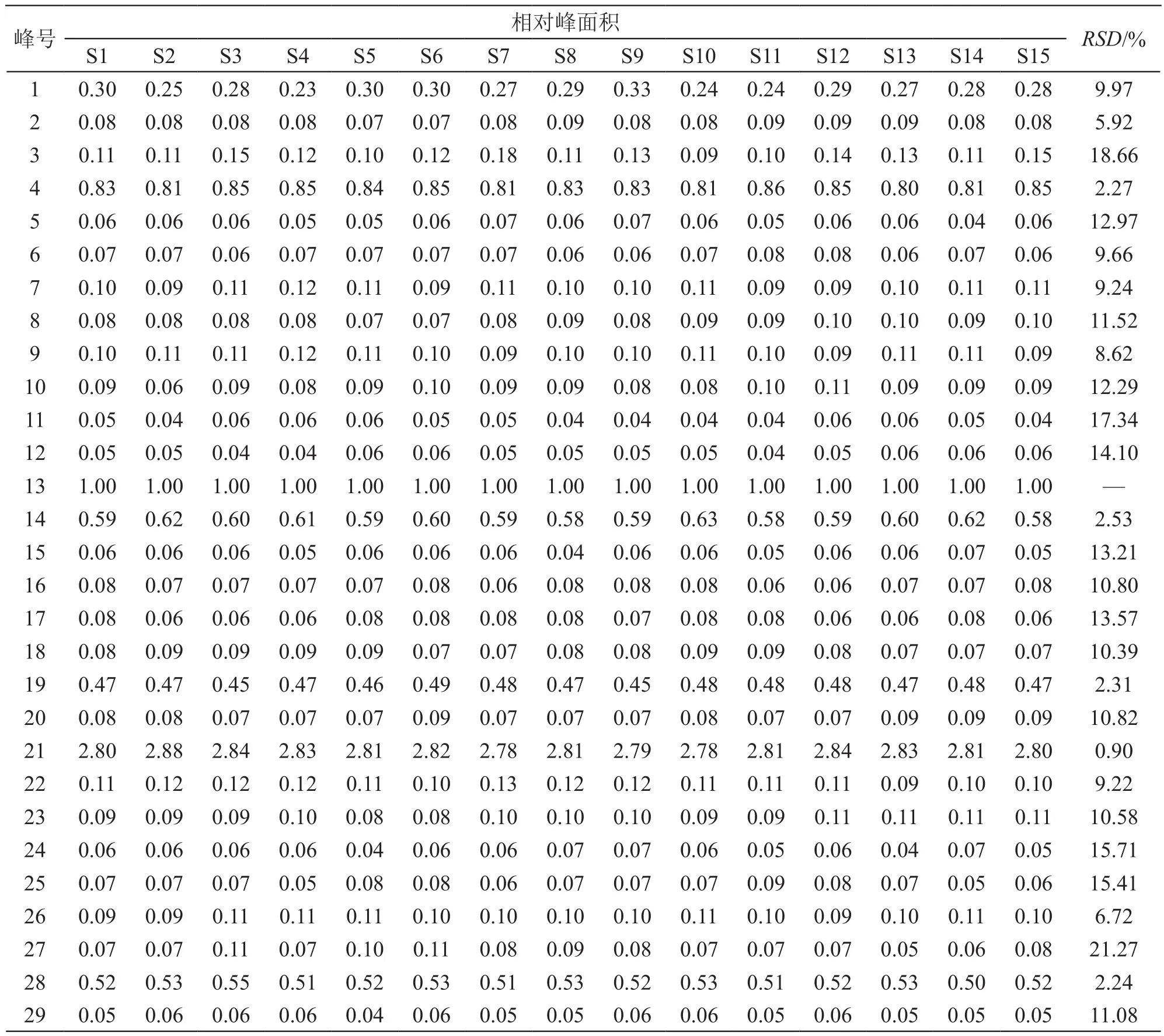
表1 15批加味香连丸共有峰相对峰面积

图2 混合对照品溶液HPLC图谱
2.5 加味香连丸中11个成分含量测定
2.5.1 色谱条件 同2.1项下色谱条件。
2.5.2 混合对照品溶液的制备 按2.2项下方法制备。
2.5.3 供试品溶液的制备 按2.3项下方法制备。
2.5.4 线性关系考察 分别精密吸取混合对照品溶液0.2,1,2,4,8,10 ml,置10 ml量瓶,用70 %甲醇溶液定容至刻度,摇匀,经0.45 μm微孔滤膜滤过,按2.1项下色谱条件进样分析,记录色谱图。以峰面积(y)为纵坐标,以样品浓度(x,μg/ml)为横坐标进行线性回归,回归方程见表2。
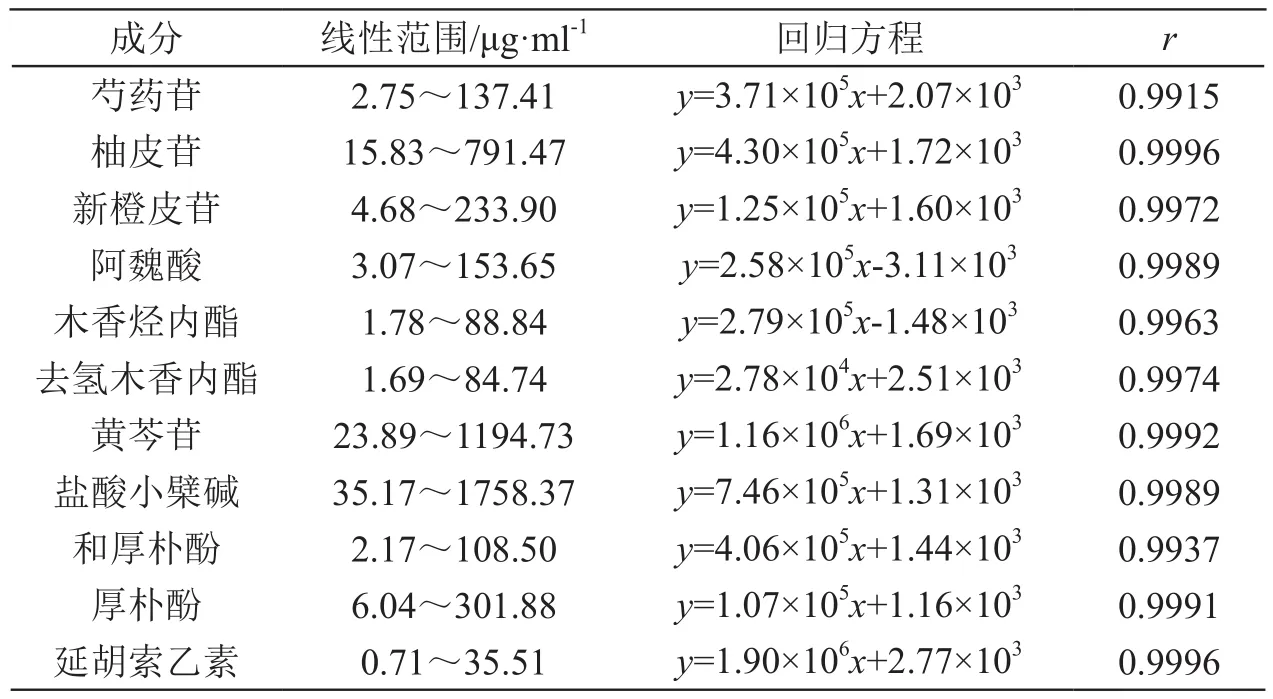
表2 回归方程和线性范围
2.5.5 精密度试验 取加味香连丸供试品(批号:2080882),按2.3项下方法制备供试品溶液,按2.1项下色谱条件连续进样6针,记录色谱图及11个化合物峰面积。结果芍药苷、柚皮苷、新橙皮苷、阿魏酸、木香烃内酯、去氢木香内酯、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素峰面积RSD分别为0.96 %,1.12 %,0.88 %,1.36 %,1.41 %,1.22 %,1.57 %,0.69 %,1.01 %,1.24 %,1.18 %(n=6),说明仪器精密度良好。
2.5.6 稳定性试验 取加味香连丸供试品(批号:2080882),按2.3项下方法制备供试品溶液,室温条件放置0,4,8,16,20,24 h,按2.1项下色谱条件进样分析,记录色谱图及11个化合物峰面积。结果芍药苷、柚皮苷、新橙皮苷、阿魏酸、木香烃内酯、去氢木香内酯、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素峰面积RSD分别为1.15 %,0.77 %,0.92 %,1.03 %,1.41 %,1.11 %,0.89 %,1.36 %,1.44 %,1.25 %,1.39 %(n=6),表明室温条件下供试品溶液在24 h内稳定。
2.5.7 重复性试验 取同一批加味香连丸供试品(批号:2080882)6份,每份约1.0 g,精密称定,按2.3项下方法制备供试品溶液,按2.1项下色谱条件进样分析,记录色谱图及11个化合物峰面积。结果芍药苷、柚皮苷、新橙皮苷、阿魏酸、木香烃内酯、去氢木香内酯、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素平均含量分别为1.041,5.996,1.772,1.163,0.673,0.642,9.051,13.321,0.822,2.287,0.269 mg/g,RSD分别为1.16 %,0.55 %,0.34 %,0.61 %,0.77 %,0.59 %,1.15 %,0.39 %,0.62 %,0.74 %,0.26 %(n=6),表明该方法重复性良好。
2.5.8 加样回收试验 取已知含量的加味香连丸供试品9份,研细,每份取约0.5 g,精密称定,置100 ml具塞锥形瓶中,分成3组,分别精密加入含芍药苷(0.208 mg/ml)、柚皮苷(1.201 mg/ml)、新橙皮苷(0.355 mg/ml)、阿魏酸(0.233 mg/ml)、木香烃内酯(0.135 mg/ml)、去氢木香内酯(0.129 mg/ml)、黄芩苷(1.813 mg/ml)、盐酸小檗碱(2.668 mg/ml)、和厚朴酚(0.165 mg/ml)、厚朴酚(0.458 mg/ml)、延胡索乙素(0.054 mg/ml)混合对照品溶液3,2.5,2 ml,按2.3项下方法制备,按2.1项下色谱条件分析,记录色谱图及峰面积,计算11个化合物的平均加样回收率及RSD值。结果芍药苷、柚皮苷、新橙皮苷、阿魏酸、木香烃内酯、去氢木香内酯、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素平均加样回收率分别为102.3 %,96.2 %,101.6 %,102.7 %,100.8 %,98.4 %,101.9 %,102.5 %,98.2 %,103.7 %,102.9 %,RSD分别为0.55 %,2.29 %,1.07 %,0.84 %,1.01 %,1.28 %,1.61 %,0.72 %,0.91 %,0.72 %,1.49 %(n=9)。具体数据见表3。

表3 加样回收结果(n=9)
2.5.9 样品含量测定 取15批加味香连丸供试品,精密称定,按2.3项下方法制备供试品溶液,按2.1项下色谱条件分析,记录色谱图及峰面积,并计算11个化合物的含量,结果见表4。
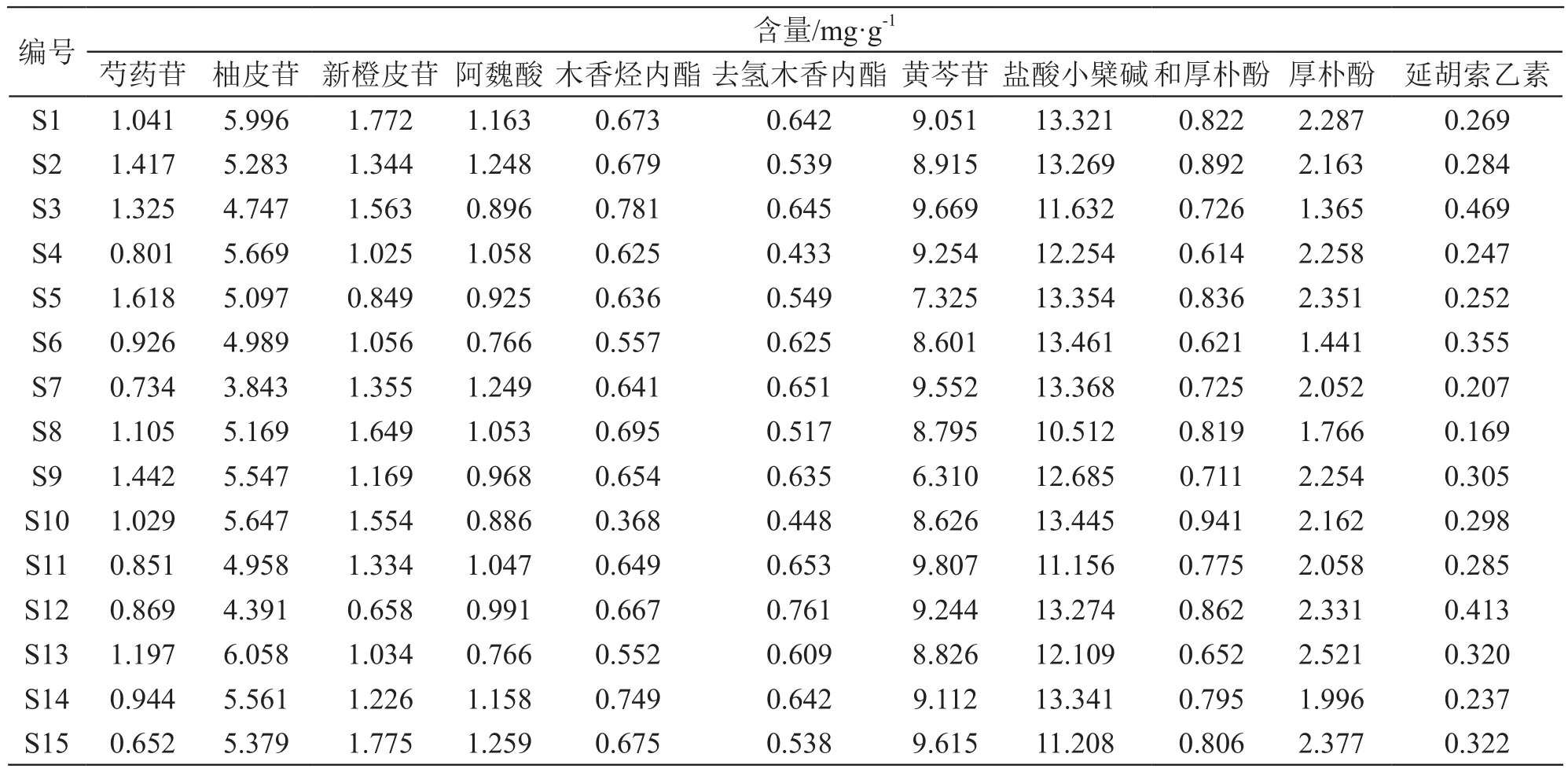
表4 样品测定结果(n=3)
3 讨论
3.1 检测波长的选择
本文采用DAD检测器,在190~400 nm全波长扫描加味香连丸供试品溶液,发现在波长280 nm处色谱峰数量较多,但芍药苷在230 nm波长处、木香烃内酯和去氢木香内酯在225 nm波长处有最大吸收,为使各成分及指纹图谱均在其最大吸收波长处进行测定,采用不同时段切换不同检测波长的方法,即0~8 min,12~29 min和32~80 min检测波长为280 nm,检测26个共有峰及柚皮苷、新橙皮苷、阿魏酸、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素,8~12 min检测波长为230 nm,检测芍药苷,29~32 min检测波长为225 nm,检测木香烃内酯和去氢木香内酯。
3.2 洗脱条件的选择
本研究首先考察了不同比例乙腈-水、甲醇-水及乙腈-甲醇-水流动相系统对加味香连丸各成分的分离效果,发现甲醇作为有机相时,供试品溶液色谱图中色谱峰较少,基线不平稳,柱压较高,而乙腈作为有机相不仅洗脱能力强、基线平稳、柱压低,且色谱峰峰形较好;水作为水相时,目标成分木香烃内酯和去氢木香内酯分离度不佳,水相更换为0.05 %磷酸水溶液后,两者可达到分离度要求,其他各色谱峰分离度也较好,基线平稳,峰形较好,指纹图谱色谱峰数量多,故选用乙腈-0.05 %磷酸作为流动相梯度洗脱系统。
3.3 供试品制备方法优化
本研究以加味香连丸指纹图谱中色谱峰数量、相邻色谱峰分离效果及11个待测化合物提取率为指标,依次对提取方式(超声提取、加热回流提取、冷浸提取)、提取溶剂(水、稀乙醇、70 %乙醇、95 %乙醇、甲醇、70 %甲醇、50 %甲醇)进行考察,结果显示70 %甲醇溶液超声提取色谱峰较多,相邻色谱峰分离效果较好,11个化合物的提取率较高;同时对70 %甲醇溶液超声提取时间(15,30,40,60 min)考察,结果显示提取时间为30 min时27个共有峰峰面积及11个化合物提取率最大,故最终确定70 %甲醇超声提取30 min作为加味香连丸供试品溶液的制备方法。
3.4 测定结果分析
15批加味香连丸相似性评价结果显示,样品与对照指纹图谱间相似度均大于0.99,说明加味香连丸批间差异较小,质量较稳定,共生成29个共有峰,通过与对照品比对指认了其中11个共有峰。11个化合物含量测定结果显示,芍药苷含量为0.652~1.618 mg/g、柚皮苷含量为3.843~6.058 mg/g、新橙皮苷含量为0.658~1.775 mg/g、阿魏酸含量为0.766~1.259 mg/g、木香烃内酯含量为0.368~0.781 mg/g、去氢木香内酯含量为0.433~0.761 mg/g、黄芩苷含量为6.310~9.807 mg/g、盐酸小檗碱含量为10.512~13.461 mg/g、和厚朴酚含量为0.614~0.941 mg/g、厚朴酚含量为1.365~2.521 mg/g、延胡索乙素含量为0.169~0.469 mg/g。根据《中国药典》标准规定加味香连丸“每1 g含黄连、黄柏以盐酸小檗碱计,不得少7.0 mg”,本研究盐酸小檗碱测定结果符合上述规定。
本研究建立了加味香连丸HPLC指纹图谱,并同时指认和测定了芍药苷、柚皮苷、新橙皮苷、阿魏酸、木香烃内酯、去氢木香内酯、黄芩苷、盐酸小檗碱、和厚朴酚、厚朴酚、延胡索乙素11个化合物的含量,方法稳定简单,能系统快速地评价该制剂的质量,对全面控制加味香连丸的质量具有指导意义。
