固体饮料中唾液酸含量的检测方法
2022-08-04李翔宇徐柳柳舒敏李慕梓李博玲汪志明
李翔宇 ,徐柳柳 ,舒敏 ,李慕梓 ,李博玲 ,汪志明 *
1. 嘉必优生物技术(武汉)股份有限公司(武汉 430073);2. 湖北省营养化学品生物合成工程技术研究中心(武汉 430223);3. 中国科学院合肥物质科学研究所(合肥 230031);4. 中国科学技术大学(合肥 230026)
唾液酸(sialic acid,SA)是一族神经氨酸的氮或氧基取代的衍生物总称,其衍生生物均为带负电的九碳糖神经氨酸[1]。SA广泛存在于哺乳动物组织中,是糖蛋白、低聚糖和糖脂的重要组成部分[2],参与细胞识别、生存、繁衍等作用[3]。脊椎动物和高等无脊椎动物体内也含有丰富的SA[4-5],如羊肉、牛肉[6]。母乳中唾液酸的含量远高于牛乳[7-8]。关于SA的功效成为各大领域研究热点。在疾病治疗方面,有研究表明,SA具有抗肿瘤[9]、抗病毒[10]等功效。对于健康人群,有研究表明,SA与神经系统发育具有相关性[11-12]。故孕妇及婴幼儿对SA的摄入量越来越受到关注。
近年来,固体饮料因其方便携带、品种丰富、易于保存等特点,受到越来越多消费者追捧。固体饮料通常分为蛋白型固体饮料和普通型固体饮料[13-14]。随着人们对食品健康与营养越来越关注,固体饮料也朝着营养化、功能化方向发展,富含功能因子的固体饮料市场需求越来越大。外源性SA的供给,可提高动物大脑中神经节苷酯的浓度,增进学习能力[15-16]。因此向固体饮料中添加SA,可以满足繁忙的消费者同时追求口感价值和营养价值。许多固体饮料生产商将其中SA含量标示出来,以此作为产品亮点。因此,开发一种可靠、简单易行的固体饮料中SA检测的标准化方法可使固体饮料中的SA检测规范化、科学化和系统化,为相关企业的研发及生产提供参考价值。
试验分别利用液相色谱法(参照GB/T 30636—2014《燕窝及其制品中唾液酸的测定 液相色谱法》)、茚三酮显色法、间苯二酚比色法、高效液相-紫外检测法及高效液相-荧光检测法检测固体饮料中SA含量,通过优化反应条件,建立一种简单易行、准确度高,不受体系中杂质干扰,可用于固体饮料中唾液酸含量检测方法,为相关研究提供理论支持及技术参考。
1 材料与方法
1.1 材料与仪器
N-乙酰神经氨酸标准品、DMB试剂、茚三酮、间苯二酚(阿拉丁);某品牌茶粉;乙腈、甲醇、磷酸、乙酸(色谱纯);低亚硫酸钠、2-巯基乙醇、硫酸铜、浓盐酸、浓硫酸、乙酸丁酯、正丁醇、硫酸铜(分析纯);超纯水。
UltiMate3000高效液相色谱仪,配荧光检测器(美国Thermo公司);高效液相Agilent1260色谱仪(配紫外检测器,美国Agilent公司);Zorbax SB-Aq色谱柱(4.6 mm×50 mm,美国Agilent公司);色谱柱Aminex HPX-87H(300 mm×7.8 mm,美国伯乐公司);ZOBAX 300色谱柱SCX阳离子交换柱(4.6 mm×250 mm,美国Agilent公司);3-18K高速冷冻干燥离心机(德国Sigma公司);HH-2智能数显恒温水浴锅(巩义市予华仪器有限责任公司);XS105分析天平(瑞士Mettler Toledo公司)。
1.2 试验方法
1.2.1 标准溶液配制
精确称取100 mg唾液酸标准品于100 mL容量瓶中,超纯水溶解并定容。将标准储备液按不同浓度梯度稀释成系列工作曲线,超纯水作试剂空白。同样品处理方法处理。
1.2.2 液相色谱法
1.2.2.1 色谱条件
流动相,乙腈-0.1%磷酸水溶液(90∶10,体积比);进样量20 μL;流速1 mL/min;柱温30±1 ℃;检测波长205 nm。
1.2.2.2 样品测定
定量称取茶粉(精确至0.000 1 g)于100 mL容量瓶中,加水至刻度处(使用温水进行溶解,需保证样品完全溶解)。按SA添加量将浓度稀释至标曲范围内,准确量取10 mL于透析袋中,在流水下透析24 h,透析后,将其放浸泡于聚乙二醇溶液中,使水分渗出。将试液转移至25 mL比色管中,并用少量水冲洗透析袋,合并试液。向其中加入乙酸,使其浓度为50%。将其置于100 ℃水浴锅中,水浴10 min,取出比色管,冷却至室温。将水解液转移至100 mL容量瓶中,用流动相定容,过膜,上机。
1.2.3 茚三酮显色法
1.2.3.1 试剂配制
茚三酮显色剂:2.5 g茚三酮溶于60 mL冰乙酸及40 mL浓盐酸中。
1.2.3.2 样品测定
定量称取茶粉(精确至0.000 1 g)于100 mL容量瓶中,加水至刻度处(使用温水进行溶解,需保证样品完全溶解)。按SA添加量将浓度稀释至标曲范围内,取2 mL溶液于比色管中,向其中加入2 mL茚三酮显色剂及2 mL冰乙酸,于100 ℃水浴锅中水浴10 min。
1.2.4 间苯二酚显色法测定茶粉中唾液酸含量
1.2.4.1 试剂配制
向50 mL容量瓶中依次加入250 μL 0.1 mol/L硫酸铜溶液、2.5 mL 4%间苯二酚和40 mL浓盐酸,用超纯水定容。摇匀避光备用。
萃取剂:乙酸丁酯和正丁醇按17∶3体积比配比。
1.2.4.2 样品测定
定量称取茶粉(精确至0.000 1 g)于100 mL容量瓶中,加水至刻度处(使用温水进行溶解,需保证样品完全溶解)。按SA添加量将浓度稀释至标曲范围内,定量取溶液于10 mL玻璃比色管中,向其中加入等量显色剂。沸水浴1 h,于冰水中冷却10 min。加入等量萃取剂,涡旋30 s后静置10 min,待分层后取上层于585 nm波长处测吸光度。
1.2.5 HPLC-UV测茶粉中唾液酸含量
1.2.5.1 色谱条件
流动相为5 mmol/L硫酸-水溶液;进样量10 μL;流速0.6 mL/min,柱温60±1 ℃;检测波长210 nm。
1.2.5.2 样品测定
定量称取茶粉(精确至0.000 1 g)于100 mL容量瓶中,加水至刻度处(使用温水进行溶解,需保证样品完全溶解)。按SA添加量将浓度稀释至标曲范围内,0.22 μm微孔滤膜过滤,供液相分析。
1.2.6 HPLC-FLD测茶粉中唾液酸含量
1.2.6.1 色谱条件
色谱条件:甲醇-0.05%乙酸水梯度洗脱,梯度条件表见表1,流动相A为0.05%乙酸水,流动相B为甲醇;进样量10 μL;流速2 mL/min,柱温30±1 ℃。
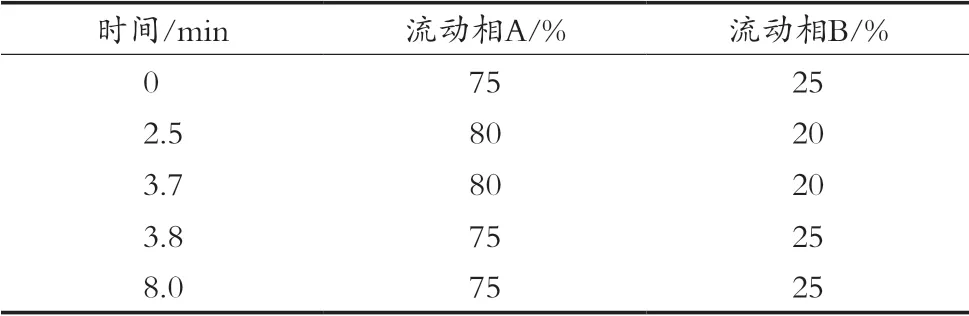
表1 色谱分离梯度条件
1.2.6.2 试剂配制
DMB试剂:向5 mL容量瓶中加入7.9 mg DMB试剂、264 μL 2-巯基乙醇、15.7 mg低亚硫酸钠及400 μL冰乙酸,最后加蒸馏水定容。
1.2.6.3 样品测定
定量称取茶粉(精确至0.000 1 g)于100 mL容量瓶中,加水至刻度处(使用温水进行溶解,需保证样品完全溶解)。按SA添加量将浓度稀释至标曲范围内,经离心处理后,取200 μL离心后的上清水解产物和相同体积的DMB溶液混合,在80 ℃下加热50 min。衍生后的样品冰浴冷却,加入400 μL水使之稀释。混匀后经0.22 μm微孔滤膜过滤,供液相分析。
2 结果与讨论
2.1 试验结果
2.1.1 液相色谱法
将标准储备液配制成质量浓度10,20,40,60,80和100 μg/mL的系列标准工作液,超纯水作试剂空白。以含量为横坐标,吸光度为纵坐标,得出回归曲线y=13 204x+1 840.9(R2=0.999 8)。结果表明,在线性范围10~100 μg/mL内,该方法具有良好线性关系。由图1可知,目标峰在10 min左右,但样品图(图2)在目标位置仅出现一点谷峰,峰面积远低于周围杂质峰,由此计算出的SA含量低于理论值的10%。故此方法不适合测茶粉中的SA,推测原因:向茶粉中添加SA的工艺多为干法添加,故体系中多为SA单体,其经透析、水解,造成大量损失。
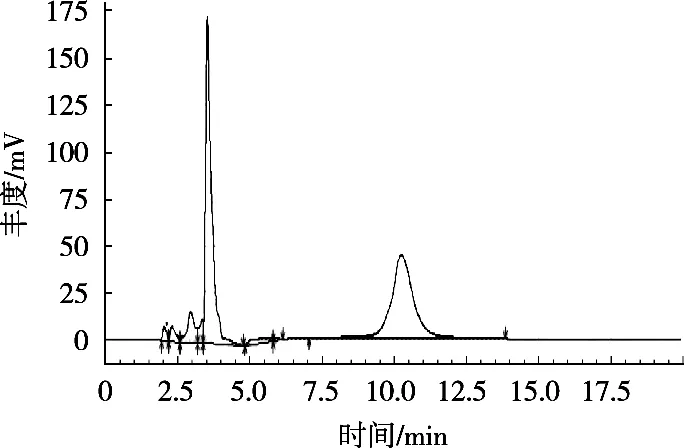
图1 SA样品液相图
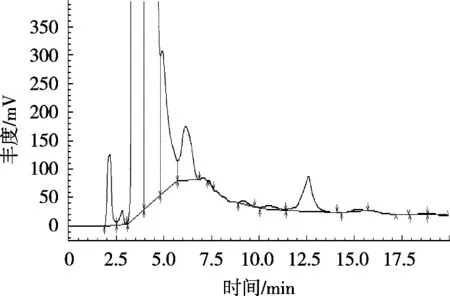
图2 茶粉液相图
2.1.2 茚三酮显色法
将标准储备液配成质量浓度5,10,20,50和100 μg/mL的系列标准工作液,用超纯水作试剂空白。以含量为横坐标,吸光度为纵坐标,得出回归曲线y=0.008 1x+0.004 3(R2=1.000 0)。结果表明,在线性范围5~100 μg/mL内,该方法具有良好线性关系。在酸性条件下,SA与茚三酮反应生成淡黄色络合物,颜色随着浓度增加而加深。研究发现,此方法测得的SA含量偏大,且结果波动较大。推测原因:茶粉体系中含有少量氨基酸,会与茚三酮发生反应,影响试验结果,试验过程中需严格控制称样量。若无空白样做本底扣除,无法计算SA添加量。
2.1.3 间苯二酚显色法
将标准储备液配成质量浓度10,20,40,80和120 μg/mL的系列标准工作液,超纯水作试剂空白。以含量为横坐标,吸光度为纵坐标,得出回归曲线y=0.008 7x-0.010 6(R2=0.999 4)。结果表明,在线性范围10~120 μg/mL内,该方法具有良好线性关系。由图3可以看出,SA标品溶液与间苯二酚反应,生产蓝色络合物,且随着浓度增加,颜色越深。而反应后茶粉溶液呈现红褐色,这可能是在酸性条件下茶粉中的酮糖与间苯二酚发生Seliwanoff反应,生成红色复合物[17],进而对试验造成严重干扰。故间苯二酚方法不适合成分复杂的茶粉中SA含量检测。由此可知,间苯二酚显色法不适合测定体系中含有酮糖的SA样品。

图3 间苯二酚法测茶粉结果图
2.1.4 HPLC-UV结果分析
将标准储备液配成质量浓度0.01,0.10,0.20,0.50和1.00 mg/mL的系列标准工作液,超纯水作试剂空白。以含量为横坐标,峰面积为纵坐标,得出回归曲线y=174.732 7x+1.315 2(R2=0.999 9),且峰型完整。结果表明,在线性范围0.01~1 mg/mL内,该方法具有良好线性关系。由图4可以看出,茶粉中SA经Aminex HPX-87H色谱柱分离后,在7.67 min处出峰,但目标峰存在拖尾、与邻近峰难分等问题,给结果分析带来很大干扰。原因可能是茶粉中含有的一些共轭基团在210 nm处也产生紫外吸收,使峰型出现拖尾,从而对检测结果有干扰,故HPLC-UV方法不适宜用来测定茶粉中SA含量。
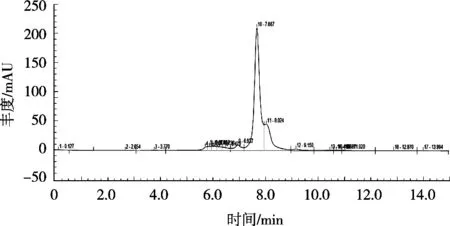
图4 茶粉HPLC-UV色谱图
2.1.5 HPLC-FLD结果分析
将标准储备液配成质量浓度0.5,1.0,2.0,5.0和10.0 μg/mL的系列标准工作液,超纯水作试剂空白。以含量为横坐标,峰面积为纵坐标,得出回归曲线y=30 383.436 7x+1 529.311(R2=0.999 9)。结果表明,在线性范围0.5~10 μg/mL内,该方法具有良好线性关系。由图5可看出,1.597 min为目标峰。茶粉中SA与DMB衍生液在酸性条件下生成具有荧光特性的衍生物,在Zorbax SB-Aq色谱柱分离后,经337 nm激发光激发后、在448 nm处产生可识别、定量的荧光信号,峰型完整,与杂质峰分离良好。可有效避免紫外检测器出现的峰拖尾现象,这与荧光检测器高选择性、高灵敏度有关。
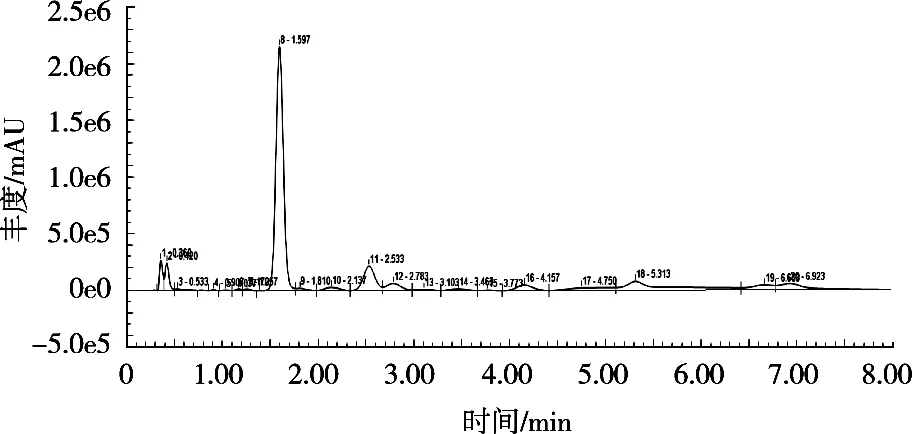
图5 茶粉HPLC-FLD色谱图
综上,HPLC-FLD检测法测定茶粉中SA含量具有良好的分离效果和较高的灵敏度。故后期可选定该方法测定固体饮料中SA含量测定。
2.2 HPLC-FLD净化条件优化
茶粉溶液中含有的蛋白质、脂肪等大分子会对后期分析产生干扰,故需要对样品溶液进行净化。分别选用Amicon Ultra-1530超滤离心管在4 000 r/min离心10 min及普通离心管在15 000 r/min下离心10 min对样品进行处理。
比较图6和图7可以得出,样品经超滤离心管离心后,峰型更完整,与周围杂质峰分离更好。经过离心管离心后的样品色谱图杂质更多,杂质峰干扰作用更大,且在目标峰旁边总存在一个较小的峰,对分析造成干扰。这说明超滤离心管对茶粉溶液的除杂作用更好,故后续试验选用超滤离心管净化样品。
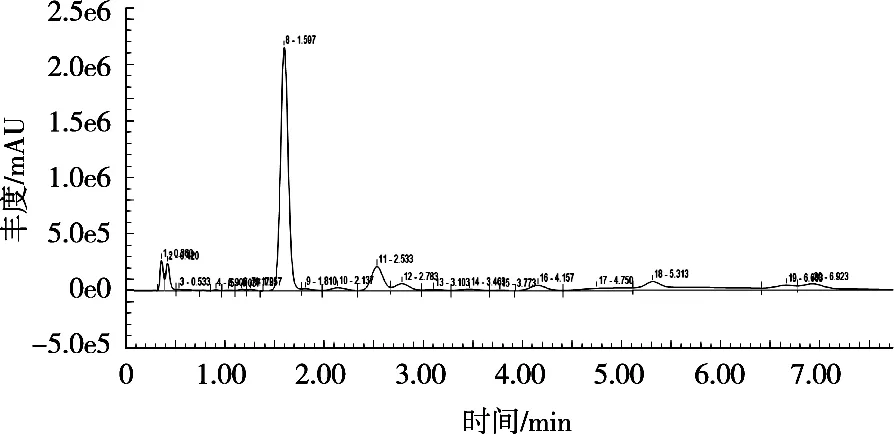
图6 超滤后茶粉HPLC-FLD色谱图
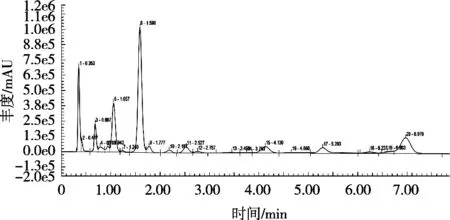
图7 离心后茶粉HPLC-FLD色谱图
2.3 检出限
以标准品色谱峰的信噪比等于3(S/N=3)对应的浓度为方法的检出限(SLOD),S/N=10对应的质量浓度为方法的定量限(SLOQ),最终确定样品中SLOD为0.2 mg/L、SLOQ为0.6 mg/L。
2.4 回收率和精密度
由于茶粉基质复杂,因此基质效应会影响检测结果的准确性,采用基质加标标准曲线定量可消除基质效应对仪器检测结果的影响。分别添加100,150和200 μL的10 μg/mL标准溶液于3种250 μL基质溶液中,补水至500 μL,进行样品处理,每个样平行6次,结果如表2所示。方法的平均加标率为101%,SRSD平均值为9.31%,表明该方法测茶粉中唾液酸含量有较好的适用性。
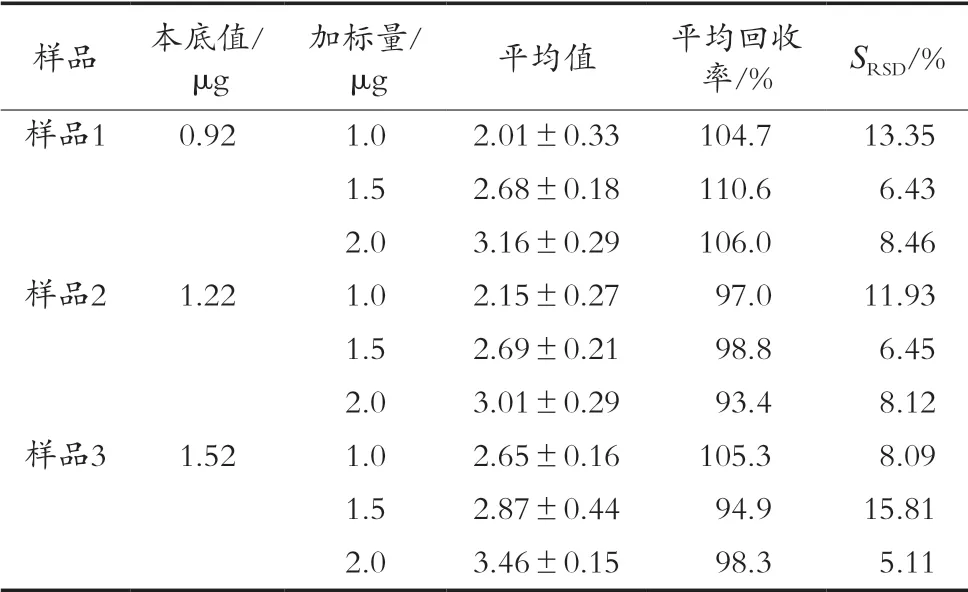
表2 样品加标回收率
3 结论
试验选定的5种方法对SA标准品测定均有很好的线性关系,但液相色谱法由于反应时间久、反应过程长,造成SA单体大量损失,故不适用于含单体SA的固体饮料的含量检测;茚三酮显色法容易受茶粉中氨基酸等成分影响,在无空白样品的情况下,无法计算SA添加量;间苯二酚法测茶粉中SA存在显色异常的问题,HPLC-UV则存在色谱峰分离度不高,杂质峰干扰等问题,故均不适合用于茶粉中SA含量检测。HPLC-FLD能克服茶粉中糖类、蛋白等杂质干扰等问题,具有重复性好、精密度高、回收率高、低检出限、易操作等优点,能准确地检测茶粉中唾液酸的含量。市面上已涌现多种多样的含SA产品,如益生菌固体饮料、果冻、美容口服液等,该方法均能满足相关产品中SA含量的检测要求,具有很好的应用前景。
