通过式固相萃取-超高效液相色谱串联质谱法测定花椒中草甘膦及代谢物残留
2022-08-04王展华张文萍柴妍熙
王展华,施 贝,张文萍,岳 超,柴妍熙
(1.浙江省食品药品检验研究院,浙江杭州 310052;2.浙江中医药大学医学技术与信息工程学院,浙江杭州 310053)
花椒是我国传统的香辛料,在我国西南地区的菜肴烹饪中占有举足轻重的作用,其衍生产品在医药、农药、防虫等方面具有重要的使用价值。近年来随着四川重庆火锅风靡全国,花椒的产销量呈递增趋势,预计到2025年,全国花椒的需求量将接近100万t。花椒种植及相关产业规模逐年扩大,但是对于可以花椒上登记使用的农药品种尚无明确规定,导致花椒产品的农药残留超标的现象时有发生。
草甘膦属于非选择类除草剂,在使用后会发生降解,其主要降解产物是氨甲基膦酸,目前对于草甘膦项目的检验主要检测草甘膦和氨甲基膦酸。随着草甘膦在全球被广泛大量应用,草甘膦及其代谢物氨甲基膦酸在环境及其生物体中富集,对人类的健康造成很大的威胁,世界卫生组织国际癌症研究机构于2015年3月20日判定,草甘膦具有遗传毒性,它可以损坏DNA,对动物致癌。在日常监测中由于草甘膦及其代谢物本身为强极性,且含膦酸基和氨基酸基两性基团,缺少发色和荧光基团,无法直接使用紫外及荧光检测器进行检测。草甘膦沸点为465.8 ℃,其代谢物氨甲基膦酸沸点为358.0 ℃,无法直接使用气相色谱进行检测。目前用于草甘膦及其代谢物的检测方法主要采用化学衍生的方法,通过降低沸点或增加发色荧光基团来改善GC和HPLC的色谱行为,提高检测响应值。
目前我国颁布的《食品安全国家标准食品中农药最大残留限量》中对谷物、油料油脂等7类食品的草甘膦限量作出了具体的规定,限量标准0.05~7.00 mg/kg;但是其中调味料中仅对叶类调味料和胡椒作出了限量规定,对花椒没有作出相应的规定。在实际风险监测中参照调味料草甘膦推荐测定方法SN/T 1923和GB/T 23750对花椒中的草甘膦残留量进行监测,由于2种方法均使用了CAX阳离子固相萃取柱进行净化,为传统的吸附-洗脱模式,试验发现,上述2种检测标准中采用净化淋洗液为水相,水相净化液需使用旋转蒸发仪进行浓缩,耗时较长,净化操作过程中需要频繁转移,操作步骤较为烦琐。笔者选取花椒为研究基质,利用亲水亲脂固相萃取柱的特点,使用通过式固相萃取技术对目标物的净化,建立通过式固相萃取-超高效液相色谱串联质谱测定花椒中草甘膦及其代谢物(氨基甲酸膦酸)残留的分析方法,以期为日常监督抽检中花椒中草甘膦及其代谢物快速检测提供依据,同时对香辛料中草甘膦及其代谢物残留检测标准制定和更新具有重要的借鉴意义。
1 材料与方法
Shimadzu LC-30AD超高效液相色谱仪(日本岛津公司);SCIEX Triple Quad5500+系统-QTRAPActivated质谱仪(美国ABSciex公司);Merck Milli-Q超纯水仪(德国默克密理博公司);Sorvall LYNX 6000 超速离心机(美国热电公司);XPE205分析天平(瑞士梅特勒公司);MultiReax多通道旋涡混合器(德国Heidolph公司);Waters ACQUITY UPLC HSS T3色谱柱(美国waters公司);Waters Oasis HLB (200 mg,6 mL) 固相萃取柱(美国沃特世公司);0.22 μm有机滤膜(上海安谱实验科技股份有限公司)。
甲醇(hypergrade for LC-MS,Merck KGaA);乙腈(hypergrade for LC-MS,Merck KGaA);丙酮(AR,国药集团化学试剂有限公司);氯甲酸-9-芴基甲酯(衍生级,纯度≥99.9%,上海安谱科学仪器有限公司);草甘膦(纯度≥99.0%,Dr.Ehrenstorfer GmbH);氨甲基膦酸(纯度≥99.0%,Dr.Ehrenstorfer GmbH);1,2-CN草甘膦(纯度≥99.8%,TRC Canada);二氯甲烷(AR,国药集团化学试剂有限公司);十水合四硼酸钠(AR,国药集团化学试剂有限公司);磷酸二氢钾(AR,国药集团化学试剂有限公司);花椒(采购于杭州联华超市)。
标准溶液的配制。
草甘膦及其代谢物标准储备液的配制。准确称取草甘膦和氨甲基膦酸各10.0 mg置于50 mL容量瓶中,用纯水溶解后定容至刻度,得到浓度为200 μg/mL的混合标准储备液。
草甘膦及其代谢物标准中间液的配制。准确量取草甘膦及其代谢物标准储备液50 μL置于10 mL容量瓶中,用纯水至刻度,得到浓度为1 mg/L的混合标准中间液。
同位素内标储备溶液的配制。准确称取1 mg的1,2-CN草甘膦置于10 mL容量瓶中,用纯水定容至10.0 mL得浓度为100 μg/mL同位素内标储备溶液。
同位素内标工作溶液的配制。取1 mL同位素内标储备溶液于10 mL容量瓶中,用纯水定容至10.0 mL得浓度为10 μg/mL同位素内标储备溶液。
样品前处理。准确称取粉碎后的花椒样品2.0 g于50 mL 塑料离心管中,同时加入100 μL同位素内标储备溶液(浓度为10 μg/mL),静置10 min,加入10 mL水,置于MultiReax多通道旋涡混合器上涡旋混合20 min,加入5 mL二氯甲烷,涡旋混合10 min,以5 000 r/min离心10 min,取水相于10 mL玻璃试管中作为待净化液。取5 mL净化液,使用Waters Oasis HLB(200 mg,6 mL)亲水亲脂固相萃取柱进行净化,弃去初始2 mL样液,收集洗脱液,待衍生。取1 mL样品洗脱液于10 mL具塞玻璃比色管中,加入200 μL 5%硼酸盐缓冲溶液,涡旋混匀。加入200 μL 1.0 g/L 氯甲酸-9-芴基甲酯-丙酮溶液,涡旋混匀。在20 ℃下放置4 h,进行衍生化反应。衍生化溶液使用0.22 μm有机滤膜过滤后进行液相色谱串联质谱测定。
标准曲线溶液的制备。分别量取草甘膦及其代谢物混合标准中间液(1 mg/L)50、100、500、1 000、2 000、5 000 μL,置于10 mL容量瓶中,用纯水定容至刻度,作为混合标准工作溶液,浓度分别为5、10、50、100、200、500 μg/L。移取标准工作溶液各1.0 mL加入同位素内标工作溶液(100 μg/mL)10 μL,按样品衍生化方法进行衍生化处理,用0.22 μm有机滤膜过滤,进行液相色谱串联质谱测定。
超高效液相色谱串联质谱条件。岛津LC-30AD超高效液相色谱系统串联Applied Biosystems Qtrap 5500三重四级杆质谱仪,包括二元泵,在线脱气,自动进样器;色谱柱为ACQUITY UPLC HSS T3 (150 mm×2.1 mm,1.8 μm);柱温为35 ℃;流动相为0.1%甲酸混合水溶液(A)和0.1%甲酸乙腈混合溶液(B);梯度洗脱程序:0~2 min,10%B,2~5 min,10%~80%B,5~8 min,80%~10%B;流速0.35 mL/min;进样量2 μL。
质谱条件:离子源为TurboV 电喷雾(ESI)离子源;电离方式为正离子;雾化电压(IS)5 500 V;离子源温度(TEM)500 ℃;雾化气压力(GS1)344.7 kPa;辅助加热气压力(GS2)344.7 kPa;碰撞室入口电压(EP)10 V;碰撞室出口电压(CXP)13 V;碰撞电压(CE)、去簇电压(DP)、定性离子对、定量离子对、化合物分子量见表1。
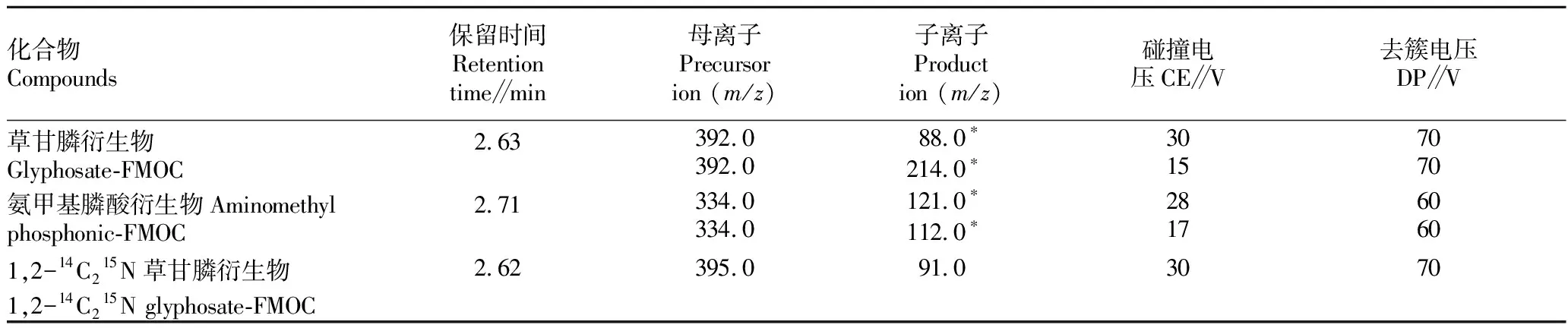
表1 质谱条件Table 1 Mass spectrometry conditions
分离色谱柱的选择。选择Waters ACQUITY UPLC HSS T3(150 mm×2.1 mm,1.8 μm)和Waters ACQUITY UPLC BEH-C色谱柱(2.1 mm×100 mm,1.7 μm)进行分离试验,结果发现,使用Waters ACQUITY UPLC HSS T3色谱柱进行分离时,草甘膦衍生物和氨甲基磷酸衍生物分离较好、峰型良好,响应优于Waters ACQUITY UPLC BEH-C色谱柱。主要原因是其C烷基键合相进行了基端封尾,提升了极性化合物的保留,能够更好分离2种物质。
液相色谱条件的优化。该研究采用Waters ACQUITY UPLC HSS T3色谱柱作为分离柱,比较了甲醇-水、乙腈-水、甲醇-水(0.1%甲酸)、乙腈-水(0.1%甲酸)4种流动相,结果表明,乙腈-水作为流动相时,由于乙腈的洗脱能力强于甲醇,对于草甘膦衍生物和氨甲基膦酸的分离度和灵敏度明显优于甲醇-水;在水相中添加0.1%甲酸可使草甘膦及其代谢物的衍生物预先在流动相中质子化,提高离子化效率,从而提高检测灵敏度。通过比较0.1%甲酸浓度对2种衍生物的影响,最终确定流动相为乙腈-水(0.1%甲酸)。在此流动相条件下各物质均能很好地分离。进一步对梯度洗脱条件进行了优化,目标物分离度良好、保留时间适中,优化后的多反应监测离子流色谱图见图1~3。
由于草甘膦和氨甲基膦酸衍生物均适合在ESI正离子模式下进行检测,使用500 μg/L的草甘膦和氨甲基膦酸标准溶液,按方法标准进行衍生化处理后,在正离子模式扫描下,根据相应物质的分子量确定一级质谱的扫描范围,分别进行一级质谱扫描找到母离子,进行二级质谱扫描确定出2~3个相应比较高的子离子,最后通过多反应检测扫描优化相应质谱参数。优化结果见表1。
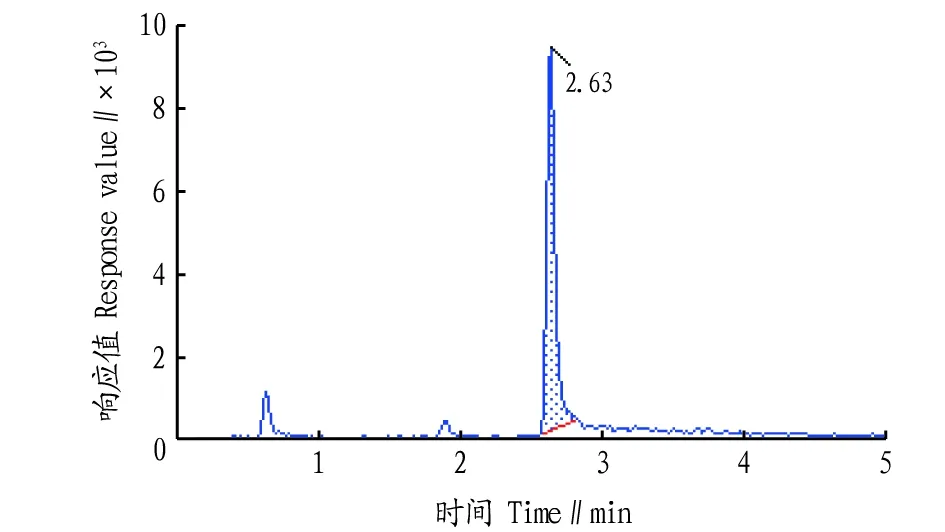
图1 1,2-14C215N草甘膦衍生物多反应监测离子流色谱图Fig.1 Multiple reaction monitoring ion chromatogram of 1,2-14C215N glyphosate-FMOC

图 2 草甘膦衍生物多反应监测离子流色谱图Fig.2 Multiple reaction monitoring ion chromatogram of glyphosate-FMOC
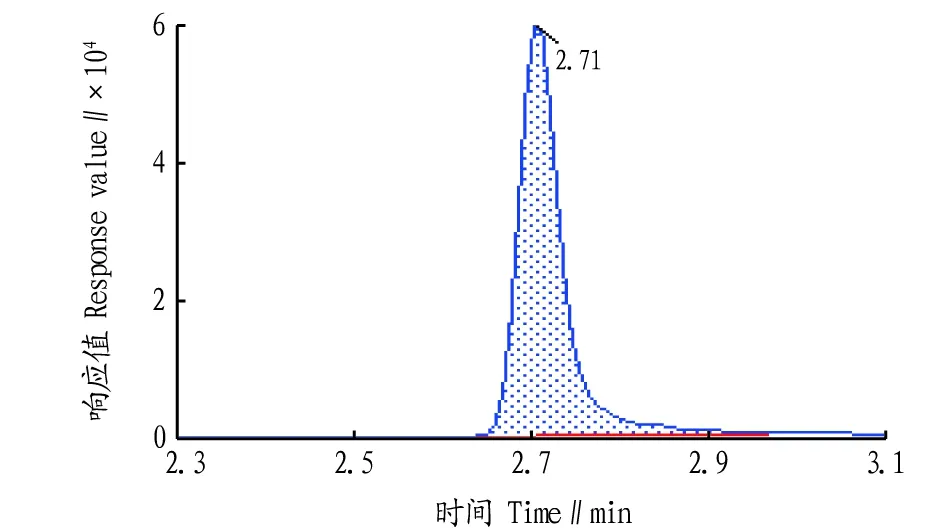
图3 氨甲基膦酸衍生物多反应监测离子流色谱图Fig.3 Multiple reaction monitoring ion chromatogram of aminomethyl phosphonic-FMOC
提取条件的优化。草甘膦及其代谢物溶于水,不溶于一般的有机溶剂,目前的国家标准和相应研究中均使用水提取溶剂。花椒样品中含有三萜、甾醇、酰胺、生物碱等化合物,其挥发油含量大于0.025 mL/g。这些大分子物质进行液相质谱分析时在对色谱柱和离子源产生污染的同时,产生较为严重的基质效应,对该研究试验结果产生较大影响。在实际研究中发现使用纯水提取的样品提取液,颜色较深,呈浑浊状态,衍生化上机后,离子化效率过低,定量限难以满足日常检验要求。该研究对纯水提取的提取液,使用5 mL二氯甲烷通过液液萃取的方式进行除脂,有效地降低了杂质过多而产生的基质效应。
固相萃取柱的选择和优化。香辛料中草甘膦测定方法SN/T 1923和GB/T 23750均使用了CAX阳离子固相萃取柱进行净化,日常监测试验发现,目标物洗脱时由于洗脱液中主要为水相从而导致浓缩时间较长、步骤复杂烦琐。在该研究,根据草甘膦及氨甲基膦酸强极性的特点,采用了Waters Oasis HLB固相萃取柱对花椒的水提取溶液进行净化。
取浓度50和200 μg/L的草甘膦及其代谢物标准混合溶液,按样品前处理方法使用Waters Oasis HLB固相萃取柱进行净化,草甘膦及其代谢物的回收率在93.1%~98.2%,结果表明采用Waters Oasis HLB固相萃取柱进行通过式净化,固相萃取柱未吸附待测物质,可以用于草甘膦及其代谢物检测的净化。
衍生条件的选择。SN/T 1923中规定使用氯甲酸-9-芴基甲酯丙酮溶液衍生需要在室温下放置过夜,使试验时间过长,在该研究中将衍生过程放置于在25 ℃恒温振荡器中,取浓度为100 mg/L的草甘膦及氨甲基膦酸标准储备液100 μL,用水定容至10 mL的1 mg/L标准中间液,添加过量衍生试剂条件下,50 r/min进行恒温振荡衍生2~8 h,每隔1 h 上机检测,结果发现衍生4 h后,测得值与衍生过夜测得值相比无明显变化,说明在衍生4 h后,衍生基本完全。此次研究确定草甘膦及其代谢物衍生时间为4 h。
方法的检出限、定量限和线性范围。采用混合标准中间液配制成5~500 μg/L的系列标准工作液绘制标准工作曲线,线性回归方程及相关系数见表2。结果表明, 草甘膦及其代谢物衍生物在5~500 μg/L线性关系良好(>0.99)。
在2 g花椒空白基质样品中加入10 μL混合标准工作液(1 000 μg/L),理论加标浓度为5.0 μg/kg,按照已建立方法检测,信噪比>3为检出限,结果表明加标浓度为5.0 μg/kg时,草甘膦和氨甲基膦酸的信噪比均大于100,满足检出限信噪比>3的要求,该方法的检出限低于SN/T 1932规定的标准(50.0 μg/kg)要求,可满足日常监督检测的需求。

表2 草甘膦和氨甲基膦酸的线性方程、决定系数、检出限Table 2 Linear equations, determination coefficients and detection limits of glyphosate and aminomethyl phosphonic acid
方法的回收率和精密度。分别选取了5、50和200 μg/kg 3个浓度梯度水平对空白样品进行加标试验,每个浓度水平测定6次,计算加标回收率和RSD值,检验结果如表3所示。结果表明,草甘膦及氨甲基膦酸的回收率分别为87.4%~96.4%、88.7%~97.1%,RSD分别为3.3%~4.2%、2.5%~4.1%,表明该方法稳定、可靠。
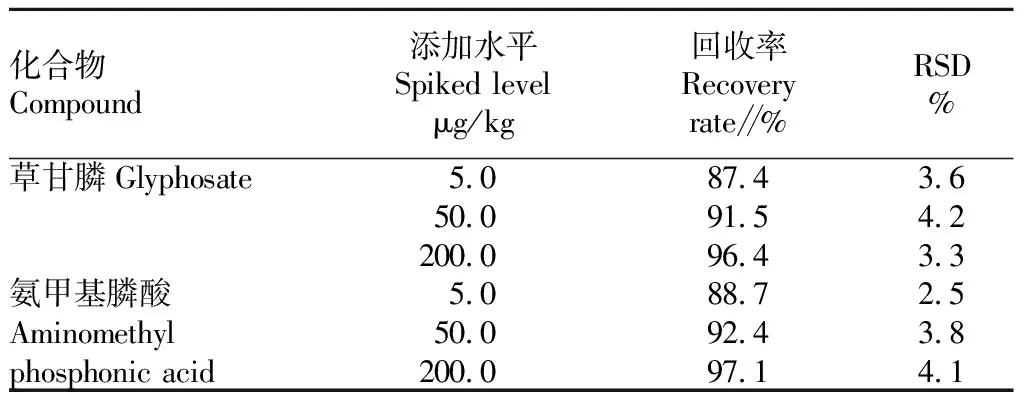
表3 草甘膦和氨甲基膦酸的加标回收率及其RSD(n=6)Table 3 Spiked recoveries and RSD of glyphosate and aminomethyl phosphonic acid
应用该方法分析了市场上采购的10份花椒样品,其中1批次样品花椒样品有氨甲基膦酸检出,检出值为20.7 μg/kg。实际样品检测结果与SN/T 1923 测得结果相比没有明显差异,说明该方法能够替代SN/T 1923用于草甘膦及其代谢物的日常检测。
3 小结
该研究建立了通过式固相萃取-超高效液相色谱串联质谱测定花椒中草甘膦及代谢物残留的检测方法,方法对提取液采用通过式固相萃取的净化方式,其净化方案与国标及其他文献中吸附-洗脱模式相比较,具有操作简便、步骤简单的特点。研究结果表明,与目前的国家相关标准相比,该方法检出限更低,定量结果更为准确,分析总耗时更短,为花椒及其他香辛料中草甘膦和氨甲基膦酸残留检测提供更加准确可靠的定性筛查和定量分析方法,也为香辛料的食品安全监管及质量控制提供技术支撑。
