Mo掺杂Co团簇催化生长单壁碳管的结构调控∗
2022-08-02张川川段海明
张川川,段海明
(新疆大学 物理科学与技术学院,新疆 乌鲁木齐 830017)
0 引言
碳纳米材料是目前纳米科技前沿领域中研究热点之一,其中对于碳纳米管[1−3]的研究始终是一个中心课题.由于其具有高强度、低密度、中空等优异的物理及化学特性,在复合材料、储氢[4−6]、催化等领域具有巨大的潜在应用价值[7−9].单壁碳纳米管(Single-Walled Carbon Nanotubes,SWCNTs)[10]因其对称性(手征性)的不同而具有不同的电导特性(金属性或半导体性)[11−14],这使得单壁碳纳米管在纳米电子器件等领域中有着极其重要的科学研究意义和应用价值,如电子电路集成[7,15]、化学和生物传感器[16−17]及单分子检测[18]等.
尽管实验上已经能够实现碳纳米管的批量生成,但直到目前,对具有单一特征(尺寸及手征性)的单壁碳纳米管的实验合成仍未能实现,目前实验合成物都包含不同尺寸和手征性的单壁碳纳米管(不具有单一的电导特征).实验合成单壁碳纳米管有多种方式,除激光灼烧法[19]和电弧放电法[20]外,最常用的是化学气相沉积法(CVD)[21−22].在CVD中,单壁碳纳米管的生长受诸多因素的影响,如温度、压强、含碳前驱体种类、催化剂组合物及催化剂载体等,其中,催化剂组合物(催化颗粒)的作用至关重要[23−25].
在采用CVD法实验合成单壁碳纳米管时通常使用3d族过渡金属作催化颗粒,如Fe[26]、Co[27]及Ni[28]等.近年来的实验研究发现,相对于单质(单元素Fe、Co及Ni)催化颗粒,它们的某些混合纳米催化颗粒(如Fe-Co[29]、Fe-Ti[30]、Fe-Mn[31]、Co-Mo[23,32]、Co-W[33]、Fe(Co、Ni)-Ru[34])具有更好的催化生长单壁碳纳米管的功能,这使得研究混合纳米催化颗粒催化生长单壁碳纳米管的催化行为细节与混合催化颗粒物性(如混合颗粒的种类、尺寸、组分等)之间的关联显得十分重要.
目前实验难以合成具有单一特征的单壁碳纳米管的一个主要原因就是对碳纳米管的催化生长机理仍没有清晰阐述.为此,实验研究与理论研究的紧密结合就显得尤为重要,而且计算机模拟研究能使我们在原子层次上加深对碳纳米管的催化生长机理的认识.当前对基于单一组分催化颗粒催化生长单壁碳纳米管有一些理论(计算)研究工作,但对基于混合催化颗粒的单壁碳纳米管的催化生长机理的理论及计算研究仍然十分少见[35].本文采用第一性原理计算方法,系统模拟研究了包含55个原子的纯Co团簇及Co-Mo混合团簇体系与不同手征性((5,5)及(10,0))单壁碳纳米管的相互作用,重点讨论分析了相较单质团簇Co55、掺杂Mo原子后混合团簇对于单壁碳纳米管的显著不同的结构调制作用.
1 计算方法和结构模型
基于密度泛函理论的第一性原理方法系统计算了Co55、Co54Mo1c及Co54Mo1t团簇与两种不同手征性单壁碳纳米管(扶手椅型(5,5)-SWCNT及锯齿型(10,0)-SWCNT)的相互作用.具体计算采用VASP(Vienna Ab initio Simulation Package)软件包[36].采用GGA-PW91的交换关联势[37],使用平面波赝势基组,原子弛豫的力收敛标准是0.01 eV/˚A,能量收敛标准是10−4eV.对于(5,5)-M55和(10,0)-M55(M55=Co55、Co54Mo1c、Co54Mo1t)体系的计算分别使用了15×15×30 ˚A和18×18×30 ˚A的盒子,以确保相邻盒子中原子间的相互作用可以忽略不计.
本文中计算单质Co55团簇为正二十面体结构,具有Ih高对称性.掺杂团簇Co54Mo1结构源于对正二十面体Co55团簇做(表面或中心)Mo原子替换.图1中给出了优化后的Co55、Co54Mo1c(Mo在中心位置)及Co54Mo1t(Mo在表面顶点位置)的几何结构、能量及对称性.
我们计算了两种不同手征性的单壁碳纳米管:扶手椅型(5,5)-SWCNT及锯齿型(10,0)-SWCNT.两种单壁碳纳米管在与以上(类)二十面体过渡金属团簇相互作用时,碳管一端与团簇相接触,另一端以H原子饱和其碳管的悬挂键.本文具体计算中(5,5)管包含了60个C原子及10个H原子、(10,0)管包含了80个C原子及10个H原子.图2给出了优化后的两类单壁碳纳米管的几何结构及能量.
此前对于单质催化颗粒与单壁碳纳米管的相互作用的第一性原理计算表明[38−39],相对于通常链接更多原子(更多配位数)的桥位吸附,顶位吸附更具有优势.本文在计算单壁碳纳米管与过渡金属团簇相互作用时,在初始相互作用构型选取上,均考虑了桥位吸附及顶位吸附两种情况.
2 结果分析与讨论
2.1 催化颗粒与扶手椅型(5,5)-SWCNT的相互作用
图3中给出了Co55、Co54Mo1c、Co54Mo1t团簇与扶手椅型(5,5)-SWCNT相互作用的稳定结构,相应结构的能量在表1中列出.从图3中可以看出,当扶手椅型(5,5)-SWCNT吸附在Co55、Co54Mo1c及Co54Mo1t团簇表面的顶位时,其优化结构相对于初始构型无明显变化(顶位吸附特征显著),当扶手椅型(5,5)-SWCNT吸附在各团簇桥位时的优化结构相对于初始构型有轻微变化,但还是保留了桥位吸附的总体特征.由于扶手椅型(5,5)-SWCNT的尺寸与团簇基底的尺寸有轻微差别,团簇碳纳米管稳定体系中吸附在团簇表面C原子的位置相对于初始构型位置都有微小变化.

表1 Co55、Co54Mo1c及Co54Mo1t团簇与扶手椅型(5,5)-SWCNT相互作用体系稳定结构的能量
分析表1中扶手椅型(5,5)-SWCNT与不同团簇基底Co55、Co54Mo1c及Co54Mo1t相互作用时稳定结构的总能量可知:扶手椅型(5,5)-SWCNT吸附在Co55和Co54Mo1c团簇的顶位比吸附在桥位时更稳定,且相较于吸附纯Co55时,顶位吸附较桥位吸附能量低1.34 eV.单原子中心位掺杂混合团簇Co54Mo1c的顶位吸附能量较桥位吸附能量显著降低(4.18 eV),此时顶位吸附的优势更加明显;然而,扶手椅型(5,5)-SWCNT吸附在混合团簇Co54Mo1t的顶位和桥位时的能量竞争关系中,相较单个Mo原子中心位掺杂情形完全相反,此时,桥位吸附具有显著的能量优势(比顶位吸附时能量低4.84 eV).
在对纯Co55团簇进行单个Mo原子替换掺杂后,混合团簇与扶手椅型(5,5)-SWCNT相互作用时,碳纳米管的吸附位置与掺杂Mo原子的位置强烈相关,随掺杂Mo原子所处位置的不同(表面顶点位或内部中心位),吸附位置明显不同.这表明可以通过调控Mo原子的掺杂位置实现对扶手椅型(5,5)-SWCNT的吸附位置的调控,这说明在Co催化颗粒中进行Mo原子掺杂能够对扶手椅型(5,5)-SWCNT的催化生长起到有益的结构调控作用.
2.2 催化颗粒与锯齿型(10,0)-SWCNT的相互作用
图4中给出了Co55、Co54Mo1c及Co54Mo1t团簇与锯齿型(10,0)-SWCNT相互作用的稳定结构,相应结构的能量在表2中列出.
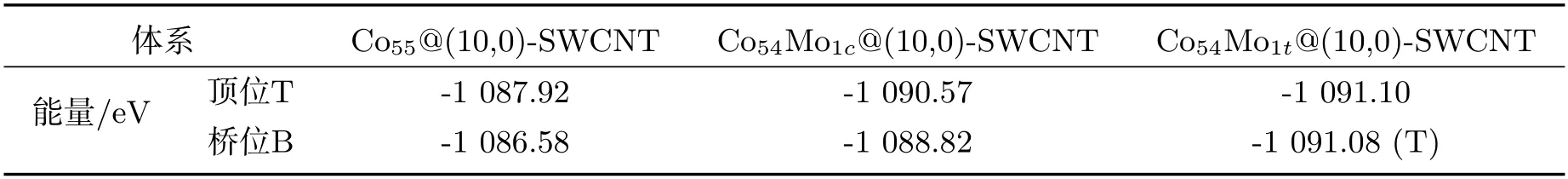
表2 Co55、Co54Mo1c及Co54Mo1t团簇与锯齿型(10,0)-SWCNT相互作用体系稳定结构的能量
分析图4及表2可知,当锯齿型(10,0)-SWCNT吸附在Co55及Co54Mo1c团簇的顶位或桥位时,其优化结构相对于初始构型均无明显变化(顶位及桥位吸附特征显著、可明显区分),且顶位吸附均较桥位吸附稳定,相对于纯团簇、混合团簇其顶位吸附较桥位吸附优势有一定增强(能量差分别为1.34 eV和1.75 eV).当锯齿型(10,0)-SWCNT吸附在Co54Mo1t团簇的顶位时,其优化结构相对于初始构型无变化(顶位吸附特征显著).而当锯齿型(10,0)-SWCNT吸附在Co54Mo1t团簇的桥位时,其优化结构相对于初始构型完全不同,体系的初始构型完全不稳定,并且优化结构过渡到顶位吸附情形(团簇与碳纳米管发生了相对旋转).可见,当锯齿型(10,0)-SWCNT与Co54Mo1t团簇相互作用时,只有顶位吸附是稳定的.
在对纯Co55团簇进行单个Mo原子替换掺杂后,无论掺杂Mo原子的位置如何,混合团簇对锯齿型(10,0)-SWCNT的顶位吸附均具有明显优势.不同之处在于:当掺杂Mo原子处于表面顶点位置时桥位吸附能够稳定存在,而当掺杂Mo原子处于内部中心位置时桥位吸附是不稳定的.这同样表明,可以通过调控Mo原子的掺杂位置实现对锯齿型(10,0)-SWCNT的吸附位置的调控.
2.3 催化颗粒对单壁碳纳米管的机理分析
由以上分析讨论可知,对纯Co55催化颗粒进行单个Mo原子替换,催化颗粒与不同手征性的单壁碳纳米管相互作用发生显著变化,掺杂团簇对于相互作用的单壁碳纳米管具有显著增强的结构调控(顶位匹配或桥位匹配)作用.这一切都来源于单个Mo原子的介入,比如,当单个Mo原子位于团簇中心位置时,相较纯Co55团簇,无论相互作用的单壁碳纳米管的手征性如何(锯齿型或扶手椅型),顶位吸附相较桥位吸附的优势均显著增强.直观地看,该情形(Mo在55原子体系中心)下,Mo原子与碳管相距较远,何以会对体系与碳管的相互作用造成如此大的影响?
图5给出了Co54Mo1c团簇与不同手征性((5,5)-SWCNT及(10,0)-SWCNT)相互作用体系最稳定构型的电子态密度(图5也给出了单独Co54Mo1c团簇的电子态密度,以虚线表示).两类相互作用体系中最稳定构型均为顶位吸附碳纳米管.分析图5可知,相较独立混合团簇Co54Mo1c的电子分布(图5中虚线部分),在混合团簇碳纳米管相互作用体系中Co、Mo的电子分布均变化不大.具体而言,含碳纳米管体系中Mo原子态密度(图5中实线)相较无碳纳米管时(图5中虚线)的明锐度(离散的尖峰)降低,这是由于含碳纳米管体系的对称性相较无纳米管的Co54Mo1c团簇明显降低,后者具有正二十面体Ih高对称性,高对称性对应于高简并度,对应于离散的尖峰现象更加突出.
对于混合团簇碳纳米管体系,由图5可知,体系各组成部分的态密度均有各自特征:(54个)Co原子态密度为相对靠近Fermi能的窄带(紧致)分布,体现出d电子主导的典型态密度分布特征;碳纳米管态密度主要由其外层s、p电子贡献,由于s、p电子相对于d电子更加离域,其态密度分布也更加宽泛.值得注意的是对两种不同手征性的碳纳米管,其相对远离Fermi能处(如处于-3∼-7 eV范围中)的电子分布较相对靠近Fermi能处(0∼-3 eV)的电子分布更加密集,这表明相对于Co催化颗粒中的电子分布(以d电子为主体),碳纳米管中的电子更倾向于靠近深能级.而掺杂Mo原子的电子分布更具独特性,尽管仍是以d电子为主,但其分布却并非以靠近Fermi能为主(这与Co明显不同),其主体电子分布均相对向较深能级扩展(如-5 eV以下Mo原子仍有明显电子分布,而Co原子电子分布几可忽略),考虑到碳纳米管中C电子的相对深能级分布(如主体分布在-5∼-7 eV),Mo原子的电子分布能使其电子与碳纳米管的电子发生更多的相互作用(更多的交叠),并且对稳定性(能量)更加有利(更靠近深能级).
可见,尽管Mo原子处在团簇中心,相距碳原子较远,但其与碳纳米管的相互作用较单个Co原子要明显强烈.我们认为,正是由于Mo原子的这种特殊的电子结构(电子分布)导致单个Mo原子掺杂Co团簇体系表现出相对于纯Co团簇的明显增强的结构调控作用.
综上所述,对于纯Co55团簇做表面顶点位的单个Mo原子替换,掺杂体系也表现出对于相互作用的单壁碳纳米管的明显增强的结构调控作用.其根本原因也来源于掺杂体系中Mo原子特殊的电子结构.图6给出了Co54Mo1t团簇与不同手征性((5,5)-SWCNT及(10,0)-SWCNT)相互作用体系最稳定构型的电子态密度(图6也给出了单独Co54Mo1t团簇的电子态密度,以虚线表示).可见如同Mo原子掺杂在中心位置,此时,Mo原子的电子分布(相较Co原子)也更加趋向于较深能级,导致Mo原子与碳纳米管有更强的相互作用,从而体现出对碳纳米管的更加明显的结构调控作用.
3 结论
采用基于密度泛函理论的第一性原理计算方法系统计算研究了纯Co55及Mo掺杂Co54Mo团簇与两类不同手征性的(5,5)-SWCNT和(10,0)-SWCNT的相互作用,分析了单壁碳纳米管对过渡金属团簇的顶位吸附和桥位吸附两种情形.对于纯Co55团簇,其与不同手征性的单壁碳纳米管相互作用时,顶位吸附均较桥位吸附稳定(能量均低约1.34 eV).而对于掺杂Mo原子混合团簇体系,其与不同手征性的单壁碳纳米管相互作用时,顶位吸附与桥位吸附的能量与掺杂Mo原子的位置密切相关:当Mo原子掺杂在团簇中心位置时,(相较纯团簇情形)顶位吸附较桥位吸附更具优势、且对于扶手椅型(5,5)-SWCNT表现得更为明显(锯齿型时能量低了1.75 eV,而扶手椅型时能量低了4.18 eV);当Mo原子掺杂在团簇表面顶点位置时,顶位吸附与桥位吸附因相互作用单壁碳纳米管的手征性的不同而表现出截然相反的能量竞争性,对于扶手椅型(5,5)-SWCNT桥位吸附较顶位吸附明显稳定(具有显著的能量优势,能量低4.84 eV),而对于锯齿型(10,0)-SWCNT顶位吸附占据完全的主导地位(桥位吸附不稳定).可见,Co-Mo混合团簇体系与单壁碳纳米管的相互作用与掺杂Mo原子在团簇中的位置及单壁碳纳米管的手征性均强烈相关.相较纯Co55团簇,Mo原子掺杂团簇对于单壁碳纳米管具有明显增强的结构调控作用,这对于单壁碳纳米管的结构可控性实验生长合成能够提供有益的借鉴.
