不同产地加工方法对香附质量的影响Δ
2022-07-29雷敬卫张维方李莹莹王心乐河南中医药大学药学院郑州450046中国中医科学院中药研究所北京00700河南省中药质量控制与评价工程技术研究中心郑州450046
纪 亮,雷敬卫,张维方,贾 豪,李莹莹,王心乐(.河南中医药大学药学院,郑州 450046;2.中国中医科学院中药研究所,北京 00700;3.河南省中药质量控制与评价工程技术研究中心,郑州 450046)
香附为常用大宗中药材,为莎草科植物莎草Cyperus rotundusL.的干燥根茎,具有疏肝解郁、调经止痛的功效,临床常用于治疗肝郁气滞、痛经等症[1],有“气病之总司、女科之主帅”之称。查阅历代炮制古籍发现,不同历史时期香附的加工方法各不相同[2],现代产地加工多为火燎后直接晒干、蒸制或煮制后晒干,广大学者对香附的研究多集中于其醋制、酒制等炮制过程和炮制品的质量评价[3-5],而未对具体产地加工方法进行系统研究。2020 年版《中国药典》(一部)规定挥发油为香附药材的质量评价指标,也有研究证实该类成分为香附的主要药效活性成分[6],但在产地加工中所使用的蒸、煮等高温处理方法势必影响香附的药效成分,其具体变化如何、对药材质量产生何种影响并未详细阐述。有学者利用高效液相色谱(high performance liquid chromatography,HPLC)指纹图谱结合化学成分含量测定,评价了香附加工炮制过程中化学成分变化与道地药材的质量差异[7-9],较好阐释了药材炮制过程中品质变化及道地药材的优选,这为香附产地加工研究提供了参考。本课题组前期产地调研发现,各地区对香附的产地加工手段不一(如河南省对香附产地加工以直接晒干为主,其他省份在保留晒干基础上,先将香附蒸、煮再于日光下晒干),加工工艺也不尽相同。同时根据本课题组前期预实验发现,不同产地加工方法所得香附样品的化学成分存在明显差异。基于此,本研究采用HPLC法建立香附的指纹图谱并进行化学计量学分析,同时采用HPLC 法测定α-香附酮、香附烯酮、木犀草素和阿魏酸的含量,多角度研究产地加工方法对香附化学成分的影响,探讨香附适宜的产地加工方法。
1 材料
1.1 主要仪器
本研究所用主要仪器有ME204E 型万分之一分析天平、AB135-S 型十万分之一分析天平[梅特勒-托利多仪器(上海)有限公司],HH-S6 型电子恒温水浴锅(巩义市予华仪器有限责任公司),1260 型HPLC 仪(美国Agilent公司),Milli-Q型超纯水处理系统[默克密理博实验室设备(上海)有限公司],KQ-700DB型数控超声波清洗器(昆山超声仪器有限公司),C21-RT2170 型电磁炉(美的集团股份有限公司)等。
1.2 主要药品与试剂
α-香附酮对照品(批号MUST-19041104)、香附烯酮对照品(批号MUST-19103001)、阿魏酸对照品(批号MUST-19032928)、木犀草素对照品(批号DST191020-032)均购于成都曼斯特生物科技有限公司,纯度均大于98%;甲醇、磷酸为色谱纯,超纯水为实验室自制。
香附药材均于2019年10月采集于河南省开封市尉氏县,经河南中医药大学药学院陈随清教授鉴定为莎草科植物莎草C.rotundusL.的干燥根茎。取香附药材5 kg,除去杂质,火燎去毛须及节上毛状物,分别采用蒸、煮、晒干等方法处理,每个方法处理3批样品。具体样品处理信息见表1。

表1 样品处理信息表
2 方法与结果
2.1 香附HPLC指纹图谱的建立
2.1.1 供试品溶液的制备 取香附药材粉末约0.5 g,精密称定,置于具塞锥形瓶中,精密加入甲醇25 mL,称定质量,超声(功率250 W,频率40 kHz,下同)处理30 min,放冷,称定质量,用甲醇补足减少的质量,摇匀,经0.22 μm滤膜滤过,即得。
2.1.2 混合对照品溶液的制备 精密称取α-香附酮和香附烯酮对照品适量,置于25 mL 量瓶中,加甲醇溶解并定容,得到上述2种成分质量浓度分别为0.277、0.278 mg/mL的混合对照品溶液。
2.1.3 色谱条件 以Venusil C18(4.6 mm×250 mm,5 μm)为色谱柱,以甲醇(A)-水(B)为流动相进行梯度洗脱(0~10 min,40%A→48%A;10~50 min,48%A→59%A;50~70 min,59%A;70~100 min,59%A→70%A;100~110 min,70%A;110~120 min,70%A→80%A);采用二极管阵列检测器,检测波长为254 nm;柱温为30 ℃;进样量为20µL;流速为1.0 mL/min。
2.1.4 精密度试验 取样品(编号A5-1),精密称定,按“2.1.1”项下方法制备供试品溶液,再按“2.1.3”项下色谱条件连续进样测定6次,以香附烯酮为参照峰,计算各共有峰的相对峰面积和相对保留时间。结果显示,各共有峰相对保留时间的RSD均小于2.00%(n=6),相对峰面积的RSD均小于3.00%(n=6),表明仪器精密度良好。
2.1.5 重复性试验 取样品(编号A5-1),精密称定,按“2.1.1”项下方法平行制备6 份供试品溶液,再按“2.1.3”项下色谱条件进样测定,以香附烯酮为参照峰,计算各共有峰的相对峰面积和相对保留时间。结果显示,各共有峰相对保留时间的RSD均小于2.00%(n=6),相对峰面积的RSD 均小于3.00%(n=6),表明方法重复性良好。
2.1.6 稳定性试验 取样品(编号A5-1),精密称定,按“2.1.1”项下方法制备供试品溶液,分别在室温下放置0、2、4、8、12、24 h 时按“2.1.3”项下色谱条件进样测定,以香附烯酮为参照峰,计算各共有峰的相对峰面积和相对保留时间。结果显示,各共有峰相对保留时间的RSD均小于2.00%(n=6),相对峰面积的RSD 均小于3.00%(n=6),表明供试品溶液在室温下放置24 h内稳定。
2.1.7 指纹图谱的建立、共有峰指认及相似度评价 取27批采用蒸、煮、晒干等方法处理的样品,按“2.1.1”项下方法制备供试品溶液,再按“2.1.3”项下色谱条件进样测定,记录色谱图。将色谱数据导入《中药色谱指纹图谱相似度评价软件(2012版)》,以Z-1样品图谱为参照(因其色谱峰峰形较好,峰信号强度适中),设定时间窗宽度为0.1 min,采用中位数法,经多点校正和全峰匹配后生成27 批样品的HPLC 叠加指纹图谱和对照指纹图谱R(图1)。结果显示,27批样品有22个共有峰,其中16号和20号共有峰分别是香附烯酮和α-香附酮(混合对照品溶液的HPLC图见图2)。将各样品与对照指纹图谱进行相似度评价,结果显示,27 批样品的相似度均大于0.9。结果见表2。

图1 香附的HPLC叠加指纹图谱及对照指纹图谱(R)
图2 混合对照品溶液的HPLC图

表2 香附HPLC指纹图谱与对照指纹图谱的相似度评价结果
2.2 化学计量学分析
2.2.1 聚类分析 将22 个共有峰峰面积进行标准化处理,采用Origin 2017软件绘制聚类分析(cluster analysis,CA)的热图,结果见图3。由图3可知,27批样品可聚为2类:B4-1~B4-3、B8-1~B8-3、B12-1~B12-3(即煮制4、8、12 min)香附样品聚为一类,其余样品聚为一类。
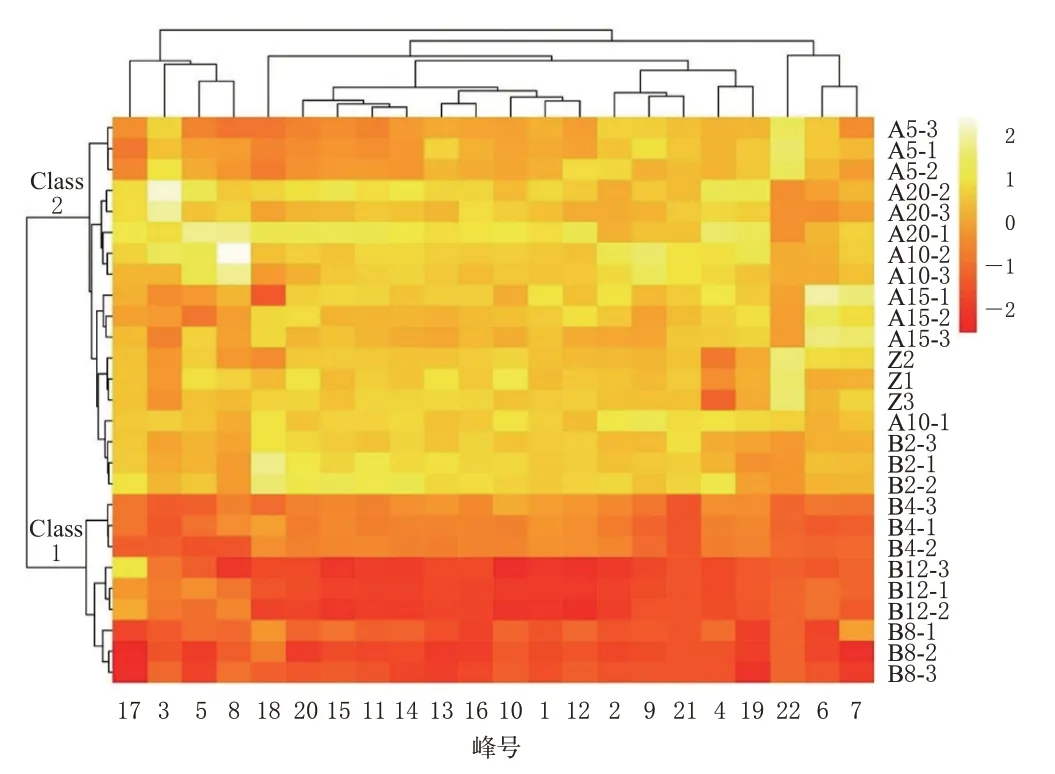
图3 27批香附样品的CA热图
2.2.2 主成分分析 主成分分析(principal components analysis,PCA)是从多个数值变量之间的相互关系入手,利用降维思想将多个变量化为少数几个互不相关的综合变量的统计方法,通常表示为原始变量的某种线性组合,能够反映原始变量的绝大部分信息[10]。运用SPSS 21.0 软件对22 个共有峰峰面积标准化处理后进行PCA。结果显示,主成分特征值大于1 的因子有3 个,3个主成分的特征值分别为17.087、1.146和1.111,贡献率分别为77.670%、5.207%和5.051%。利用相应数据计算其主成分得分(principal components score,PCS),并以此为基础计算样品的综合得分(composite score,CS),CS 越高,表示样品质量越好[11]。CS=(PCS1×77.670+PCS2×5.207+PCS3×5.051)/87.928(权重系数分别为3个主成分的贡献率,PCS代表主成分得分)。
从CS折线图(图4)可以看出,A10-1、A10-2、A10-3、A15-1、A15-2 和B2-2 样品CS 较其他样品更高,其次是“Z”样品和“A20”样品。由此推测,蒸制10、15 min和煮制2 min的香附样品质量最佳。同时蒸制处理对样品的质量影响较小,样品的质量接近;而煮制处理对样品的质量影响较大,煮制2 min后香附CS急剧下降。

图4 主成分模型的CS图
2.2.3 偏最小二乘法-判别分析 为了筛选对香附质量影响较大的成分,采用SIMCA14.1软件进行偏最小二乘法-判别分析(partial least squares discriminant analysis,PLS-DA),建立PLS-DA模型。结果显示,得分散点图中不同样品各自沿轴分开,具有明显差异性。对模型进行置换验证,R2拟合直线在Y轴截距小于0.3,说明所建模型可靠;Q2拟合直线在Y轴截距小于0.05,说明所建模型不存在过度拟合,可用于分析各样品间的差异[12]。PLS-DA得分图、模型验证图和变量重要性投影(variable important projection,VIP)图见图5。筛选VIP 值>1 的共有峰[13],找到对整体模型贡献率高于平均值的共有峰,分别为20 号峰(α-香附酮)、16 号峰(香附烯酮)、22号峰、17 号峰和21 号峰,提示对应的5 种成分可能是影响香附质量的标志性差异成分。
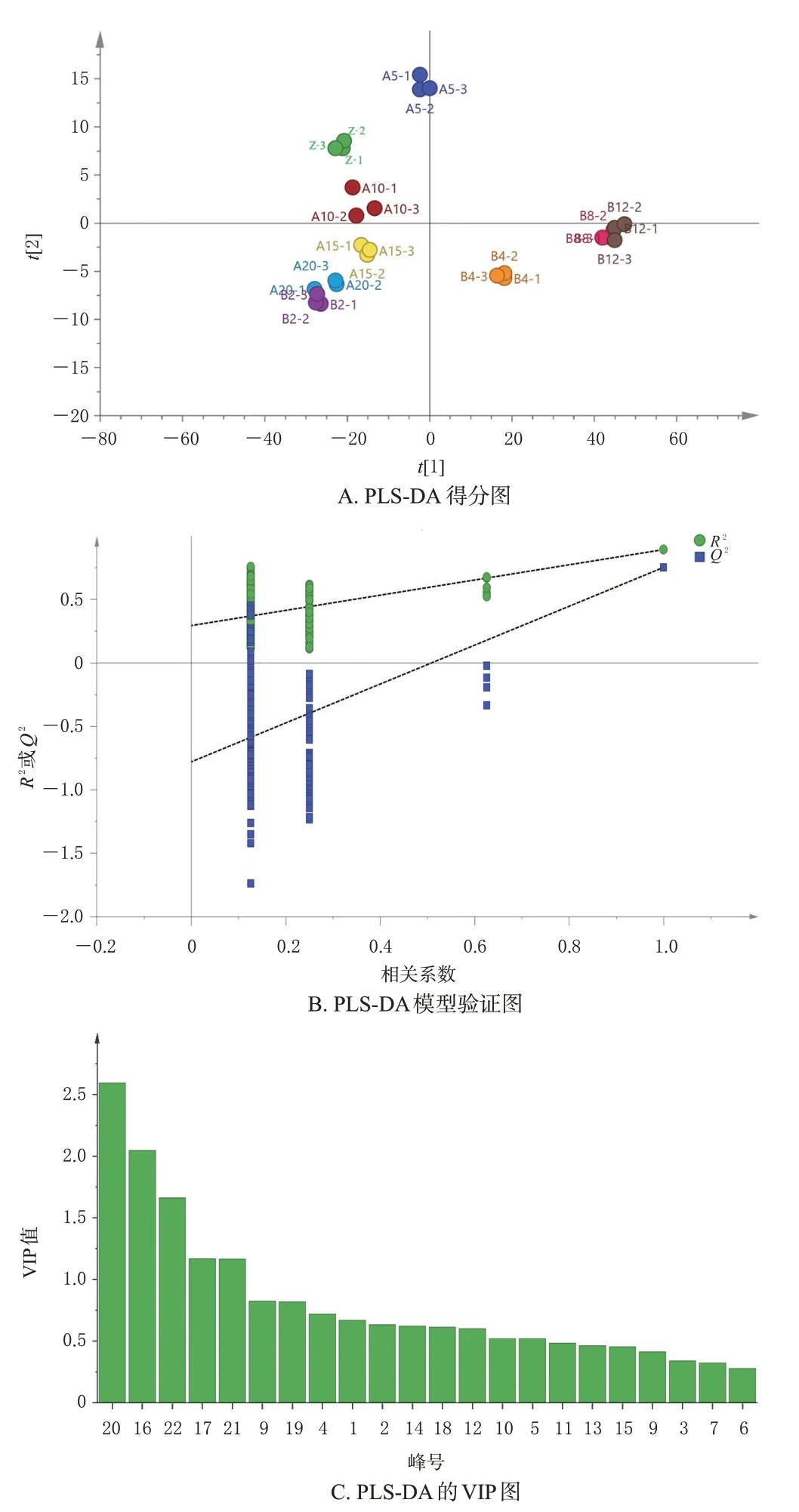
图5 PLS-DA结果图
2.3 香附中4种化学成分含量的测定
2.3.1 供试品溶液的制备(1)测定α-香附酮与香附烯酮供试品溶液的制备:同“2.1.1”项。(2)测定木犀草素供试品溶液的制备:取香附药材粉末约3.0 g,精密称定,置于具塞锥形瓶中,精密加入甲醇50 mL,称定质量,超声处理30 min,放冷,称定质量,用甲醇补足减失的质量,摇匀,滤过,取35 mL续滤液水浴蒸干,加甲醇溶解并定容至5 mL,经0.22 μm 微孔滤膜滤过,即得。(3)测定阿魏酸供试品溶液的制备:取香附药材粉末约2.0 g,精密称定,置于具塞锥形瓶中,精密加入70%甲醇40 mL,称定质量,80 ℃水浴回流30 min,放冷,称定质量,用70%甲醇补足减失的质量,摇匀,滤过,取30 mL续滤液水浴蒸干,加70%甲醇溶解并定容至5 mL,经0.22 μm 微孔滤膜滤过,即得。
2.3.2 对照品溶液的制备(1)α-香附酮与香附烯酮的混合对照品溶液的制备:同“2.1.2”项。(2)木犀草素对照品溶液的制备:精密称取木犀草素对照品适量,置于25 mL量瓶中,加甲醇定容,得质量浓度为0.59 mg/mL的木犀草素对照品溶液。(3)阿魏酸对照品溶液的制备:精密称取阿魏酸对照品适量,置于25 mL 量瓶中,加70%甲醇定容,得质量浓度为0.54 mg/mL 的阿魏酸对照品溶液。
2.3.3 色谱条件(1)α-香附酮与香附烯酮的色谱条件:以Venusil C18(4.6 mm×250 mm,5 μm)为色谱柱,以甲醇-水(68∶32,V/V)为流动相;检测波长为242 nm;流速为1.0 mL/min;柱温为30 ℃;进样量为20 μL。(2)木犀草素的色谱条件:以Venusil C18(4.6 mm×250 mm,5 μm)为色谱柱,以甲醇-0.1%磷酸溶液(45∶55,V/V)为流动相;检测波长为345 nm;流速为1.0 mL/min;柱温为25 ℃;进样量为20 μL。(3)阿魏酸的色谱条件:以Venusil C18(4.6 mm×250 mm,5 μm)为色谱柱,以甲醇-0.2%醋酸溶液(25∶75,V/V)为流动相;检测波长为320 nm;流速为1.0 mL/min;柱温为30 ℃;进样量为20 μL。在上述各色谱条件下,各目标色谱峰的分离度均大于1.5。结果见图6。
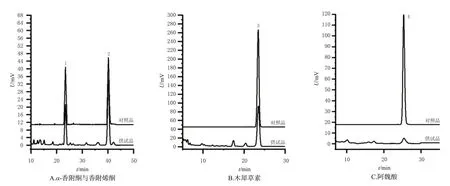
图6 供试品溶液与对照品溶液的HPLC图
2.3.4 线性关系考察(1)α-香附酮与香附烯酮标准曲线的建立:分别精密吸取“2.3.2(1)”项下α-香附酮与香附烯酮的混合对照品溶液2.0、1.5、1.0、0.5、0.1、0.006 mL,置于5 mL量瓶中,用甲醇定容,按“2.3.3(1)”项下色谱条件进样,记录峰面积。(2)木犀草素标准曲线的建立:分别精密吸取“2.3.2(2)”项下木犀草素对照品溶液2.0、1.5、1.0、0.5、0.2、0.04 mL,置于5 mL量瓶中,用甲醇定容,按“2.3.3(2)”项下色谱条件进样,记录峰面积。(3)阿魏酸标准曲线的建立:分别精密吸取“2.3.2(3)”项下阿魏酸对照品溶液3.0、2.0、1.0、0.4、0.2、0.016 mL,置于10 mL量瓶中,用70%甲醇定容,按“2.3.3(3)”项下色谱条件进样,记录峰面积。以对照品质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归。结果见表3。
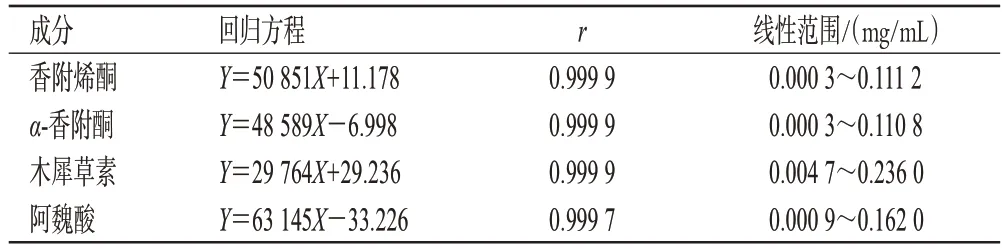
表3 香附烯酮等成分的回归方程与线性范围
2.3.5 定量限与检测限考察 精密吸取“2.3.2”项下对照品溶液适量,用甲醇或70%甲醇倍比稀释,按“2.3.3”项下色谱条件进样测定,以信噪比10∶1对应的质量浓度作为定量限,以信噪比3∶1 对应的质量浓度作为检测限。结果显示,香附烯酮、α-香附酮、木犀草素和阿魏酸的定量限分别为0.000 3、0.000 3、0.004 7、0.000 9 mg/mL,检测限分别为0.0001、0.0001、0.001 4、0.000 2 mg/mL。
2.3.6 精密度试验 取样品(编号A5-1),精密称定,按“2.3.1”项下方法制备供试品溶液,再按“2.3.3”项下色谱条件连续进样测定6次,记录峰面积。结果显示,香附烯酮、α-香附酮、木犀草素和阿魏酸峰面积的RSD 分别为0.48%、0.74%、0.81%、2.15%(n=6),表明仪器精密度良好。
2.3.7 稳定性试验 取样品(编号A5-1),精密称定,按“2.3.1”项下方法制备供试品溶液,分别在室温下放置0、2、4、6、8、12、24 h 时按“2.3.3”项下色谱条件进样测定,记录峰面积。结果显示,香附烯酮、α-香附酮、木犀草素和阿魏酸峰面积的RSD 分别为0.22%、0.65%、0.82%、2.51%(n=7),表明供试品溶液在室温下放置24 h 内稳定性较好。
2.3.8 重复性试验 取样品(编号A5-1),精密称定,共6份,按“2.3.1”项下方法制备供试品溶液,再按“2.3.3”项下色谱条件进样测定,记录峰面积并按标准曲线法计算各成分含量。结果显示,香附烯酮、α-香附酮、木犀草素和阿魏酸含量的RSD 分别为2.17%、0.52%、0.55%、2.12%(n=6),表明方法重复性较好。
2.3.9 加样回收率试验 分别精密称取已知含量的样品(编号A5-1)9份,分为3组,分别按样品中成分含量的50%、100%和150%加入对应成分的单一对照品,按“2.3.1”项下方法制备供试品溶液,再按“2.3.3”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果显示,香附烯酮、α-香附酮、木犀草素和阿魏酸的平均加样回收率分别为98.25%、98.98%、97.32%、98.08%(n=9),RSD分别为1.36%、1.24%、1.78%、1.52%(n=9),表明方法准确度良好。
2.3.10 样品含量测定 取27批样品,按“2.3.1”项下方法制备供试品溶液,再按“2.3.3”项下色谱条件进样测定,记录峰面积并按标准曲线法计算各成分的含量,每批样品测定1 次,取均值表示处理方法相同样品的测定结果。结果显示,与Z样品比较,B4、B8和B12样品中α-香附酮、香附烯酮和木犀草素的含量明显降低,A15、A20样品中香附烯酮和木犀草素含量也明显降低,表明煮制对香附中化学成分含量影响较大,在煮制2 min 后有较多成分消失。结果见表4。

表4 不同产地加工方法香附中4种化学成分的含量测定结果(mg/g)
3 讨论
香附作为大宗药材,临床应用十分广泛,可与多种中药配伍使用。现行2020年版《中国药典》(一部)中收载的以香附为原料的中药制剂高达100 多种,市场对香附的需求呈增加趋势。香附多为野生,广泛生长于田野地头,属于农副产品。本课题组实地调研发现,农民因生产条件有限且药材收益不高,往往不进行复杂的加工处理。故本研究充分模拟原始产地采收及后续加工方法,经蒸、煮后自然晒干,对不同产地加工香附的HPLC指纹图谱及多成分含量进行对比分析。结果显示,蒸制10、15 min和煮制2 min的香附样品质量最佳,其次是直接晒干和蒸制20 min处理的香附样品;CA结果显示,煮制4、8、12 min 香附样品聚为一类,与其余样品明显分开;PLS-DA 结果表明,指纹图谱中VIP 值大于1 的共有峰有5 个,分别为20 号峰(α-香附酮)、16 号峰(香附烯酮)、22号峰、17号峰和21号峰。
HPLC 指纹图谱作为中药研究体系中的重要手段,虽然在提供较多成分信息的同时,可以供研究者以宏观整体角度分析中药样品成分,提供全面的化学成分信息,但无法说明具体的成分含量值差异和变化。本研究发现,α-香附酮和香附烯酮可能为影响香附质量的标志性差异成分,但香附化学成分复杂,具有多种药理作用,现代研究在证实α-香附酮和香附烯酮为香附抗痛经及镇痛的物质基础的同时,也表明香附中黄酮类成分木犀草素和苯丙素类成分阿魏酸具有显著的抗菌、抗炎、抗血小板聚集、抗氧化等药理活性[14-16],具有较大的研究价值。由于HPLC 指纹图谱研究时的提取方法和色谱条件存在局限性,使得指纹图谱未能对木犀草素和阿魏酸进行综合表征,故本研究在对指标性成分的定量分析中,综合考虑其研究价值,选择α-香附酮、香附烯酮、木犀草素和阿魏酸为定量分析的评价指标。结果显示,不同产地加工方法对香附化学成分含量影响较大,随着蒸、煮时间的增加,α-香附酮、香附烯酮和木犀草素的含量均呈明显的降低趋势,且煮制处理对其影响极大。
综上所述,蒸制10、15 min和煮制2 min的香附样品质量最佳,与药典规定的“蒸透”和“略煮”的记载一致。查阅文献发现,产地加工采取蒸煮处理可达到杀酶保苷、便于净制、消杀虫卵等目的,其中含淀粉、糖分多的药材不易干燥,须先经蒸、煮或烫法处理,以便干燥及加工[17]。而香附药材淀粉含量极高,且糖类含量达10%以上,鲜药材摊晒过程较长,推测“略煮”及“蒸透”处理的目的是使药材更易干燥,减少在干燥过程中有效成分损失。但由于蒸煮所处的高温环境会影响药材的化学成分,故蒸、煮时间过长也会影响药材质量,而煮制处理较之蒸制处理更为猛烈,在煮制2 min 后样品CS 急剧下降,所以在使用煮制处理香附药材时,必须严格控制水煮时间,以防止过度水煮造成药材有效成分的大幅减少。
