小鼠雄性生殖干细胞转录组分析揭示成熟精原干细胞特征
2022-07-27郭彦杨乐乐戚华宇
郭彦,杨乐乐,戚华宇
小鼠雄性生殖干细胞转录组分析揭示成熟精原干细胞特征
郭彦1,杨乐乐2,戚华宇2
1. 中国科学技术大学,生命科学与医学部生命科学学院医药生物技术系,合肥 230026 2. 中国科学院广州生物医药与健康研究院,细胞谱系与发育研究中心,广州 510530
精原干细胞(spermatogonial stem cells, SSCs)是成年动物睾丸中的成体干细胞,具有自我更新与分化的能力。小鼠()精原干细胞来源于胚胎期的原始生殖细胞(primodial germ cells, PGCs),小鼠出生前原始生殖细胞处于有丝分裂静止状态,出生后恢复增殖并由曲细精管中央迁移至管壁基质,建立稳定的精原干细胞克隆。成熟小鼠的精原干细胞周期性地启动精子发生以维持雄性动物长期稳定的生殖能力。精原干细胞在其建立和成熟后是否具有特征上的差异目前尚不清楚。本研究在前期建立的不同年龄小鼠精原干细胞(表达多能性基因编码的OCT4)转录组数据基础上,对小鼠新生期(出生后3天)、幼年期(出生后7天)和成熟期(2~3月龄)精原干细胞的基因表达差异进行了生物信息学分析,包括差异表达基因(differentially expression genes, DEGs)的筛选、DEGs编码的蛋白相互作用网络(protein-protein interaction, PPI)的建立、功能聚类富集(Gene Ontology, GO)和通路分析(Kyoto Encyclopedia of Genes and Genomes, KEGG),以及使用基于GO、KEGG和HALLMARK的基因集富集分析(gene set enrichment analysis, GSEA)。结果显示,OCT4阳性精原干细胞在小鼠新生期、幼年期和成熟期存在大量差异表达基因,所编码的蛋白主要生物学功能集中在生物合成和能量代谢、免疫反应、细胞连接和迁移以及细胞分化等方面。精原干细胞细胞膜成分的显著变化可能影响精原干细胞的超敏反应、细胞间相互作用以及对细胞外环境因子的应答反应。在能量代谢方式上,随着年龄的增加,OCT4阳性精原干细胞逐渐从线粒体氧化磷酸化作用转变为糖酵解作用,同时也显著减少了细胞内核糖体形成相关基因的转录。这些结果为进一步研究雄性生殖干细胞形成和成熟的调控机制提供了新的思路。
精原干细胞;OCT4;转录组;差异表达基因
在全世界范围内,不孕不育症影响着越来越多的人群。据统计,在育龄夫妇中,8%~12%的夫妇存在不孕不育问题[1]。其中,男性因素造成的不育疾病约占50%[2]。在雄性性腺(睾丸)中,精原干细胞是支持精子发生的成体干细胞。在正常生理环境下,精原干细胞通过自我更新和分化产生阶段性的精原细胞和精母细胞,继而进入减数分裂产生单倍体精子细胞,后者通过细胞形态发生形成精子[3~5]。精原干细胞的自体移植可以帮助恢复和治疗部分由于生精障碍造成的不育症。在哺乳动物模型中的研究表明,一些失去生育能力的牛()和猴()通过精原干细胞的自体移植可以恢复睾丸组织精子发生的能力并产生有功能的精子[6~9]。精原干细胞的自体移植技术为在性成熟之前(如青春期) 由于癌症等因素造成的生殖能力丧失人群提供了一种可以保留生育能力的可能性。
精原干细胞的临床应用目前面临许多尚未解决的问题。例如,精原干细胞还没有被认可的、单一的标记分子,这对精原干细胞的研究和应用都造成一定的阻碍。对小鼠的研究表明,同种异体移植的、分化启动的未分化精原细胞在不育受体睾丸组织中可以产生长期生精的精原干细胞群落,表明睾丸内发挥干细胞功能的细胞数量或大于已知干细胞数量[10~12]。因此,有学者提出应该基于它们是否具有基本的干细胞功能,即自我更新和多能性,来识别精原干细胞[13]。目前,鉴定和评估精原干细胞功能的主要方法是观察细胞移植入曲细精管后在受体中是否具有形成克隆并长期产生精子的能力[14]。由于移植后的精原干细胞需从曲细精管管腔转移至基底膜(归巢),因此对精原干细胞表面蛋白和迁移调控的研究对提高和改善细胞移植效率具有一定意义。研究表明,小鼠中精原干细胞移植后稳定克隆形成率约为10%,在小鼠等常用动物模型中,归巢的精确效率尚不明确[15,16]。另一方面,由于动物体内精原干细胞数量的稀少,发展不同物种中长期稳定的体外培养体系,也有助于获得足够数量的、可用于移植的精原干细胞。
虽然精原干细胞的体外培养在小鼠、大鼠()、山羊()和人()等物种[17]已取得了一定的成功,但是伴随体外培养产生的遗传不稳定性会导致移植细胞诱发癌症的发生[3]。同时长期的体外培养可能会造成精原干细胞多能性下降、端粒缩短以致无法无限增殖[17]。目前,用于体外长期培养的精原干细胞多来源于年幼动物的睾丸,成年后动物个体的精原干细胞相比而言仍较难以在体外长期维持[18,19]。通过对幼年期和成熟期精原干细胞的转录组数据进行生物信息学分析,或许可以为精原干细胞体外培养技术的改良提供参考。
目前普遍认为,由于成年小鼠的睾丸中体细胞的成熟以及不同发育阶段生精细胞的增长,形成了与幼年期小鼠不同的精原干细胞生态位(精原干细胞周围可以维持其干细胞特性,并决定其自我更新或分化命运的微环境)[20,21],进而引起精原干细胞性质的变化[22]。已有的研究表明,衰老的精原干细胞分化过程受到严重阻碍,尤其是在表观遗传水平,许多促分化基因的甲基化减少,分化信号通路中的基因甲基化分布异常[23]。动物个体从新生到逐渐发育成熟,直至衰老失去生育能力,这一漫长的发育过程是否会伴随着精原干细胞自身特征的不断变化,目前新生和成熟动物体内精原干细胞间的差异研究相对较少。为探究不同年龄阶段小鼠精原干细胞的特征变化,本研究在前期建立的不同年龄小鼠精原干细胞(表达多能性基因编码的OCT4)转录组数据的基础上,对小鼠新生期(出生后3天)、幼年期(出生后7天)和成熟期(2~3月龄)OCT4阳性精原干细胞的转录组数据进行了比较分析,旨在从转录组水平对小鼠个体生长发育过程中的精原干细胞变化获得新的认识,为精原干细胞的研究提供新的方向和思路,为促进精原干细胞在临床上的广泛应用提供参考。
1 材料与方法
1.1 原始数据过滤和表达矩阵有效性检查
出生后3天(3 days-post-partum, 3-dpp)、出生后7天(7-dpp)和2~3月龄(2~3-M)的雄性OG2转基因小鼠(表达处于启动子调节下的GFP)的精原干细胞的流式分选、RNA提取和cDNA文库建立,在本实验室的另一项工作中完成。其中,通过流式分选出的、用于文库建立的OCT4阳性精原干细胞的纯度达到95%以上[24]。质量合格的样品文库送至安诺优达基因科技(北京)有限公司,使用Illumina高通量测序平台进行双端测序。3个时间点测序样品各设置3个生物学重复,一共9个测序样品。在本研究中,3-dpp组代表新生期精原干细胞前体细胞,7-dpp组代表幼年期精原干细胞,2~3-M组代表成熟期精原干细胞。
原始RNA测序数据(raw reads)去除任意一端含有测序接头的序列和低质量序列,得到高质量的有效数据(clean reads)。使用HISAT2[25]将各样品的有效数据与小鼠参考基因组进行比对。为防止表达量低的基因数据对整体测序数据分析结果的影响,分析中去除了同一基因原始读数丰度(raw read counts)大于1且样本个数小于6的基因数据。然后利用R软件的DESeq2[26]中归一化算法(中位数比率法)对所有基因的原始读数丰度进行归一化,检验常见基因(如管家基因)在每个样本中的表达量是否正常,并检查表达矩阵中每个样本的基因表达量和表达矩阵数据的有效性。
1.2 基因的差异表达分析
基于归一化后的样本RNA-seq数据,对3组基因表达数据进行组间的差异基因分析,即7-dpp3-dpp (幼年期精原干细胞与新生期精原干细胞前体细胞比较)、2~3-M7-dpp (成熟期精原干细胞与幼年期精原干细胞比较)、2~3-M3-dpp (成熟期精原干细胞与新生期精原干细胞前体细胞比较)。使用R语言中DESeq2[26]进行基因差异表达分析。由于不同阶段细胞内的基因表达整体水平存在差异,需要为不同组间的表达差异设置合适的log2FoldChange值。依据整体数据通过统计学公式:
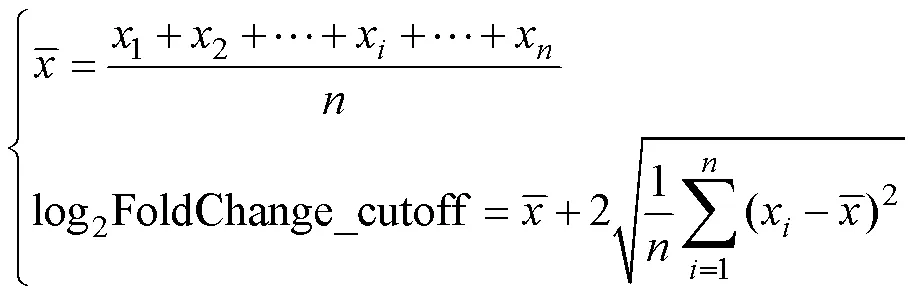
计算适合的log2FoldChange值[27],确定差异表达基因列表值<0.05,|log2FoldChange|7-dpp vs 3-dpp> 1.278,|log2FoldChange|2~3-M vs 7-dpp>1.755,|log2FoldChange|2~3-M vs 3-dpp>2.226。其中,x是某一基因log2FoldChange的绝对值。
1.3 qRT-PCR验证
利用qRT-PCR方法对筛选出的差异表达基因进行表达量的验证。随机选取7个差异表达基因,根据NCBI (https://www.ncbi.nlm.nih.gov/)提供的mRNA序列,使用在线引物设计工具Primer BLAST (https:// www.ncbi.nlm.nih.gov/tools/primer-blast/)设计引物(附表1)。以9个样品的cDNA为模板,在Bio-Rad CFX96 Touch实时定量PCR仪上使用两步法进行qRT-PCR扩增。反应条件:95℃ 30 s;95℃ 5 s,60℃ 20 s,39个循环;70℃ 5 s;在70℃~92.4℃区间内以每秒上升0.4℃绘制熔解曲线。以为内参,使用2–ΔΔCt法计算目的基因的相对表达量。
1.4 蛋白–蛋白互作(PPI)网络密集连接区域分析
在STRING (Search Tool for the Retrieval of Interacting Genes, https://string-db.org/)数据库[28]中分别输入3个阶段(3-dpp、7-dpp、2~3-M) OCT4阳性精原干细胞的差异基因,并下载与差异表达蛋白存在互作关系的蛋白质信息。利用Cytoscape 3.9.0软件[29]创建蛋白质与蛋白质的互作网络。
MCODE[30]是Cytoscape软件中的一个插件,用于对整个网络进行聚类分析以便识别出密集连接的区域。通过拓扑分析获得的聚类子网络中的每个节点至少与个其他节点相连接。K核(k-core)是重要的分析参数,一个节点的特征性k核值越大,该节点所在子网络位置的结构就越稳定。设置最小k核值为2后,选择综合评分(子网络总节点数乘以总连接数与理论最大连接数的比值,即子网络总节点数与子网络密度之积)最高的前两个子网络簇作为进行功能分析的候选簇。对PPI网络进行聚类以确定在3个阶段OCT4阳性精原干细胞中重要的特征变化网络,并由DAVID(Database for Annotation, Visualization and Integrated Discovery, https://david.ncifcrf. gov/)在线数据库对位于子网络中的节点进行功能注释[31]。
1.5 基于多个基因集的基因集富集分析
使用基于GO、KEGG和HALLMARK基因集的GSEA方法对所有基因进行分析[32~35],分别获得3个发育阶段的HALLMARK功能富集结果、潜在GO功能和KEGG通路结果。用Benjamini和Hochberg方法控制错误发现率[36]。选择调整后的值小于0.05的GO和KEGG通路。利用R软件筛选出3个阶段均被富集到的潜在GO功能,并将属于同一GO功能的潜在基因组合在一起作为该条GO功能的潜在基因,然后将所有上调和所有下调的GO功能潜在基因分别进行KEGG分析。同样地,利用R软件找出成熟期OCT4阳性成熟期精原干细胞与新生期精原干细胞前体细胞以及幼年期精原干细胞均具有显著差异的潜在KEGG通路,并对潜在KEGG通路下的所有潜在基因做GO分析。
1.6 统计分析
RNA测序样本数据的处理和分析在用于统计计算和分析的R软件环境中进行。所有数据均以mean± s.e.m表示。
2 结果与分析
2.1 归一化转录数据有效性分析
鉴于对小鼠个体差异和批次效应的考虑,以及为了使组内和组间的测量数据具有可比性,在分析测序数据之前本研究对数据进行了归一化处理(见材料与方法1.1)。数据经过归一化后,各样本的整体基因表达量处于同一水平,表明样本数据间可以进行比较,不存在批次效应(图1A)。而相同细胞类型来源的总体基因表达分布从概率密度的角度是相似的(图1B)。由于管家基因通常被用做比较基因表达变化时的参照物,其表达水平往往高于细胞内其他基因的平均表达水平。与此一致,本研究中精原干细胞样品的基因平均表达量均在5以下(图1A),而和在样本中的表达量几乎都高于10 (图1C)。样品聚类分析显示,来自同一年龄组别的3个样品在空间距离上相近,且相关性高,其次是精原干细胞前体细胞和幼年期精原干细胞的空间距离相较于成熟期精原干细胞。上述结果显示,进行转录组分析的表达矩阵数据具有可靠性和可比性。
2.2 差异表达基因的筛选和表达量聚类分析
为了探究不同阶段精原干细胞之间的差异,本研究利用DESeq2进行差异基因表达分析,并筛选出组间的差异表达基因。在组间(7-dpp3-dpp、2~3-M7-dpp、2~3-M3-dpp)差异表达基因分析中,共获得948个差异表达基因。其中,与新生期精原干细胞前体细胞相比,幼年期精原干细胞中93个基因表达量上调,311个基因表达量下调(图2A);与幼年期精原干细胞相比,成熟期精原干细胞中有323个基因表达量上调,95个基因表达量下调(图2B);与新生期精原干细胞前体细胞相比,成熟期精原干细胞中有310个基因表达量上调,284个基因表达量下调(图2C)。对所有差异表达基因进行表达量的聚类分析,发现同一组别下的3个样本形成独立的簇,即3-dpp、7-dpp和2~3-M样品的3个实验重复组分别各自形成一个独立簇(图2D)。依据在不同发育阶段OCT4阳性精原干细胞中的表达变化,所有的差异表达基因可大致分为5类:1)在7-dpp时表达下调,2~3-M时表达上调的基因;2)在3-dpp和7-dpp时低表达,2~3-M时表达上调的基因;3) 3-dpp到2~3-M表达持续上调的基因;4) 3-dpp到2~3-M持续表达下调的基因;5)在3-dpp时高表达,7-dpp和2~3M低表达的基因(图2E)。差异表达基因分析表明,3个不同年龄小鼠中的OCT4阳性精原干细胞的基因表达整体水平的趋势是不同的,与新生期精原干细胞前体细胞相比,幼年期精原干细胞的变化主要由较多基因表达下调引起。但与幼年期精原干细胞相比,成熟期精原干细胞的变化主要是基因表达上调的结果。
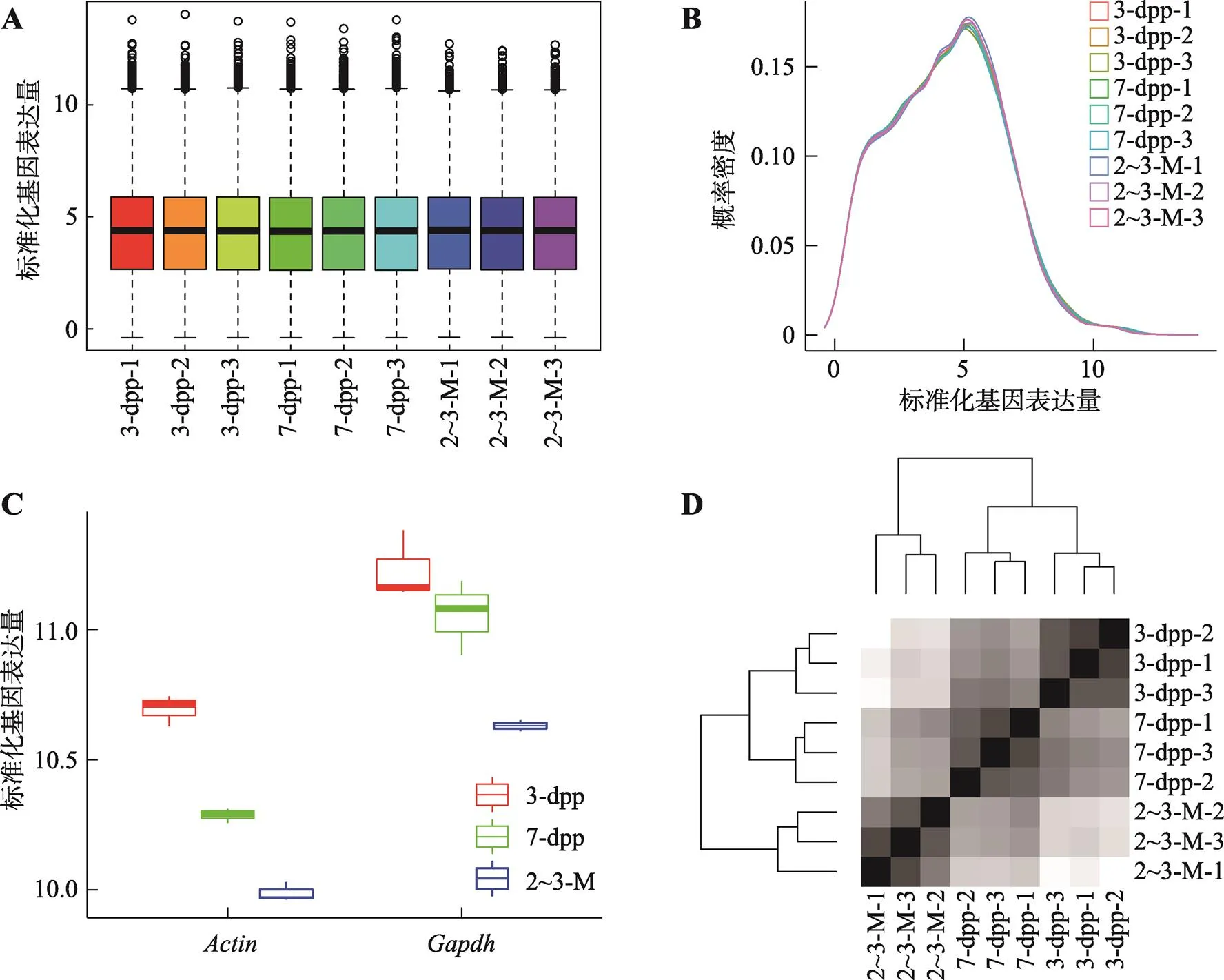
图1 归一化基因表达数据有效性分析
A:各样本所有基因在经过标准化后的整体平均表达量;B:各样本基因表达量总体分布概率密度图;C:管家基因和在各组中的标准化后表达量;D:包含所有基因的样品间相关性分析热图。
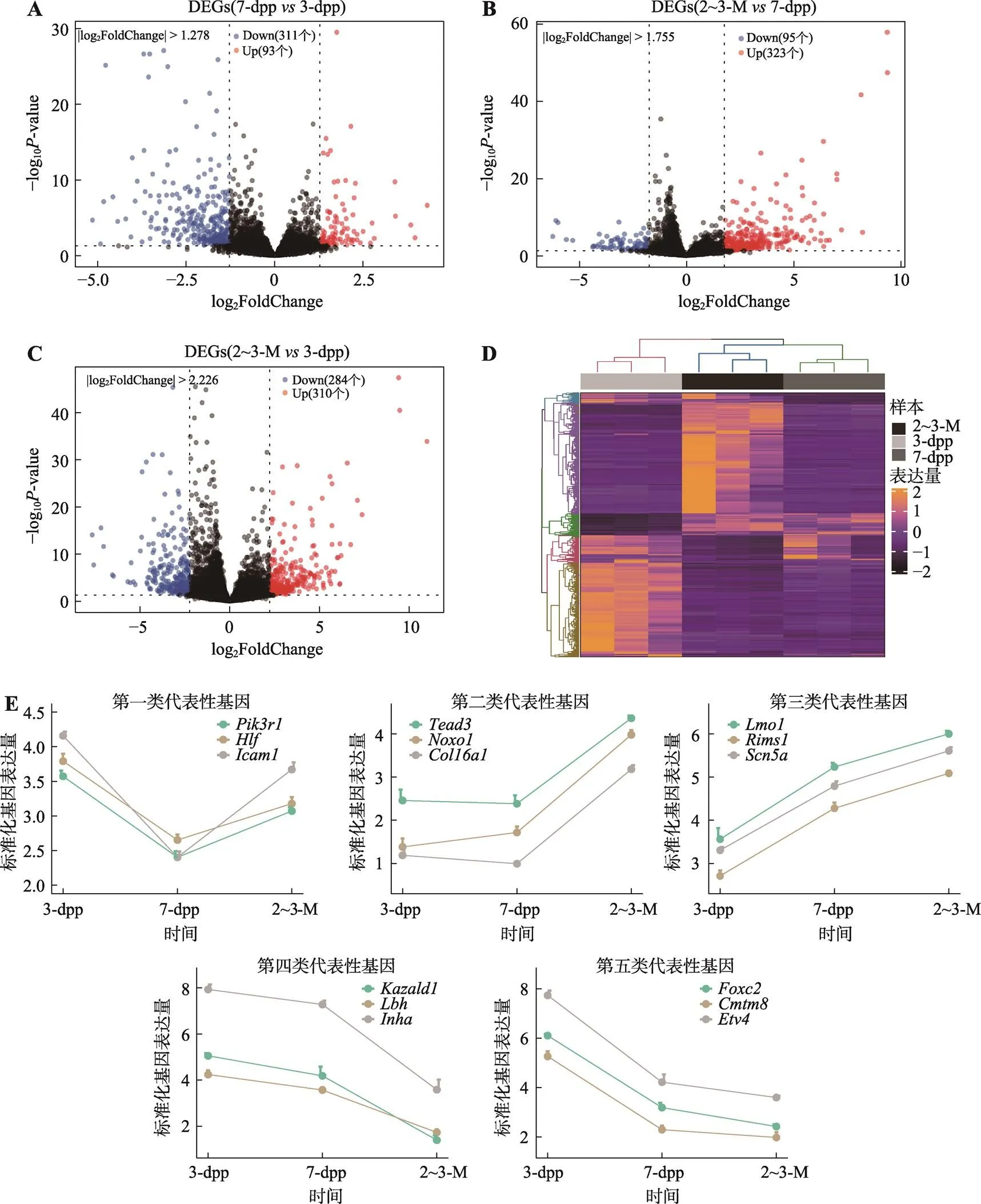
图2 差异表达基因及其表达量聚类分析
A:7-dpp3-dpp的基因差异分析结果。设置|log2FoldChange|>1.278。B:2~3-M7-dpp的基因差异分析结果。设置|log2FoldChange| > 1.755。C:2~3-M3-dpp的基因差异分析结果。设置|log2FoldChange| > 2.226。< 0.05为差异表达基因。红点表示上调差异表达基因,蓝点表示下调差异表达基因,黑点表示非差异表达基因。D:所有差异表达基因热图。E:5类表达趋势的代表性差异表达基因折线图。数据均以mean±s.e.m表示。
2.3 差异表达基因qRT-PCR验证
为了验证RNA-seq数据的真实性和差异表达基因的表达趋势,本研究利用qRT-PCR方法对随机选取的7个差异表达基因()进行了表达量测定。结果显示,所选的7个基因相对表达量曲线与RNA-seq数据呈现的表达趋势相似(图3),显示用于生物信息学分析的RNA-seq数据具有较高的可信度。
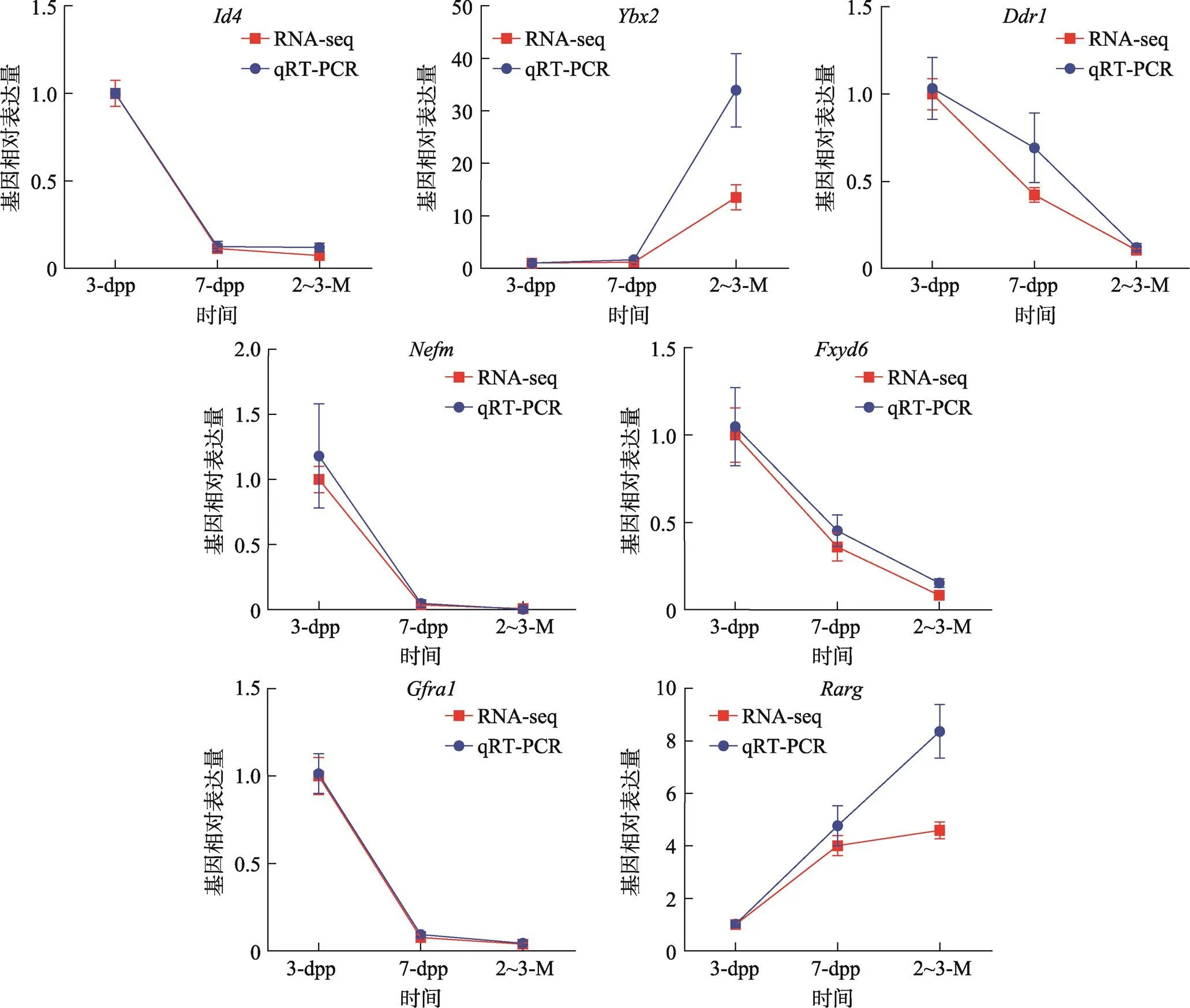
图3 随机选取的7个差异表达基因qRT-PCR验证
数据均以mean±s.e.m表示。
2.4 蛋白–蛋白互作聚类子网络构建及筛选
蛋白–蛋白互作(PPI)网络描述了蛋白质与蛋白质之间的相互作用关系,对了解细胞内蛋白质的功能和蛋白质间相互作用具有重要意义。本研究在筛选出的组间差异表达基因基础上构建了相应阶段的PPI网络。为了获得在3个阶段OCT4阳性精原干细胞间差异表达基因PPI网络中重要的网络结构及相关基因,本研究利用Cytoscape 3.9.0软件的MCODE插件对构建的差异表达基因PPI网络进行分析。新生期精原干细胞前体细胞与幼年期精原干细胞的PPI网络共聚集到14个模块簇,包含90个基因和516个互作关系(图4A)。幼年期精原干细胞与成熟期精原干细胞的差异表达基因PPI网络共聚集到15个模块簇,包含75个基因和217个互作关系(图4B)。新生期精原干细胞前体细胞与成熟期精原干细胞的差异表达基因PPI网络共聚集到17个模块簇,包含106个基因和379个互作关系(图4C)。
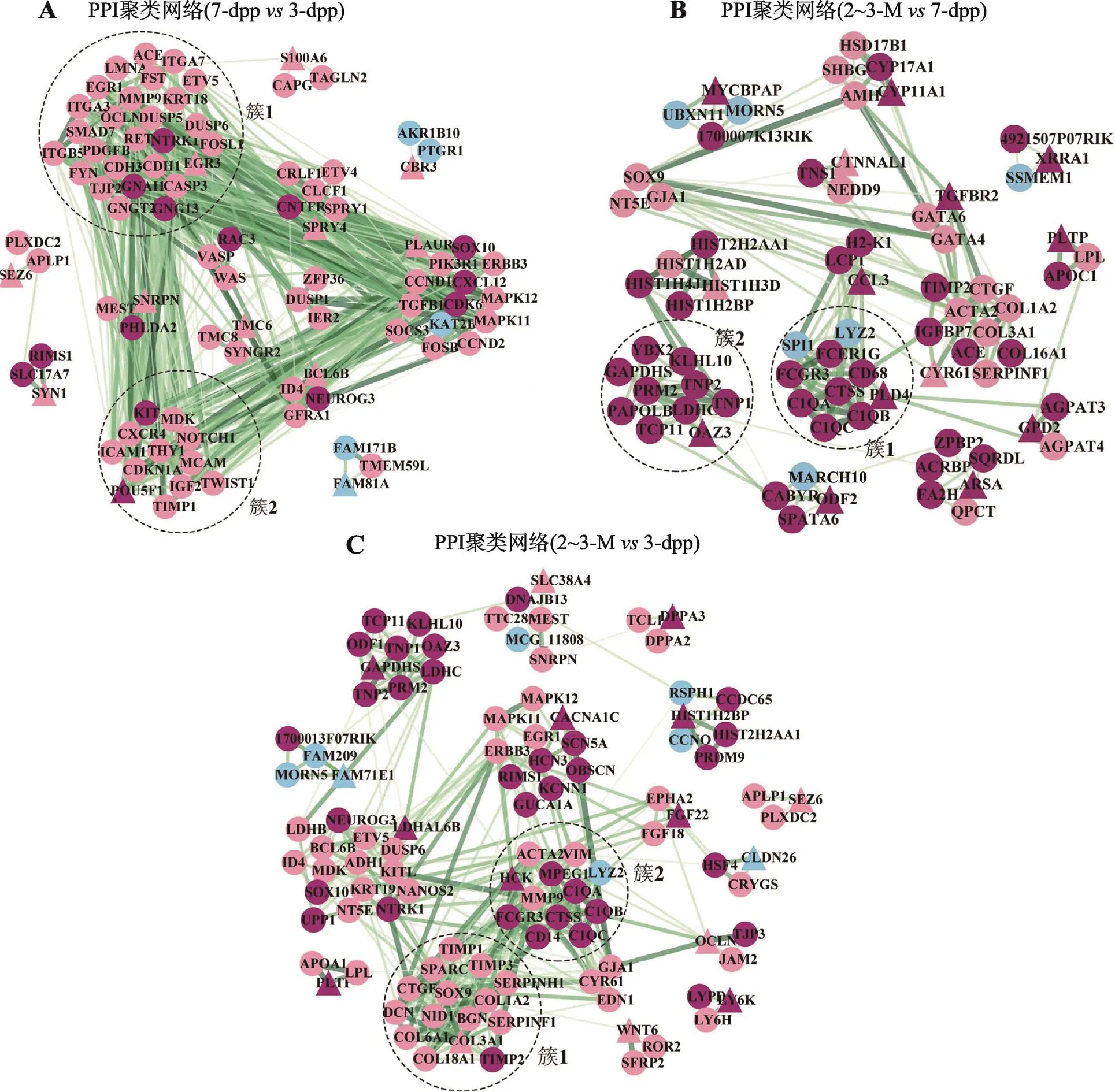
图4 蛋白–蛋白互作聚类网络簇
A:7-dpp3-dpp差异基因PPI聚类网络簇(共14个);B:2~3-M7-dpp差异基因PPI聚类网络簇(共15个);C:2~3-M3-dpp差异基因PPI聚类网络簇(共17个)。图中每个基因团是聚类分析得到的一个模块簇。虚线圈出的是综合评分最高的2个聚类网络簇。紫红色表示转录上调,粉色表示转录下调,蓝色表示差异表达基因互作节点(node),三角形为种子节点,圆圈为聚类节点,节点间结合强度越大,节点间的边(edge)颜色越深、宽度越宽。
2.5 聚类子网络功能分析
PPI综合评分(子网络总节点数与子网络密度之积)最高的两个聚类子网络(图4虚线所注),可以提示对应发育阶段OCT4阳性精原干细胞的重要蛋白相互作用特征,选择这些模块中的基因进一步分析有助于了解特定细胞中重要聚类子网络涉及的生物学功能。分别选择3个阶段中综合评分最高的两个子网络模块簇作为进行功能分析的候选簇,进行聚类子网络功能分析。如图5A所示,新生期精原干细胞前体细胞与幼年期精原干细胞相比,在幼年期精原干细胞中富集的第一个PPI网络聚类子网络基因前10条GO功能主要集中在细胞膜表面、细胞间或与细胞外基质的粘附、细胞间的连接等方面;富集的前10条KEGG通路主要与细胞粘附、细胞自我更新等相关。幼年期精原干细胞中富集的第二个聚类子网络基因前10条GO和KEGG通路则提示细胞迁移,以及通过细胞膜表面变化来改变对外界环境刺激的应答。因此,相较于新生期精原干细胞前体细胞,幼年期精原干细胞可能通过细胞表面的变化来改变对来自细胞外环境或其他细胞的信号反应,并且伴随有活跃的迁移活动。
幼年期精原干细胞与成熟期精原干细胞相比,在成熟期精原干细胞中富集的第一个聚类子网络基因前10条GO和KEGG通路均与免疫系统活动有关,尤其是超敏反应被显著正向调控(图5B)。而第二个子网络模块的富集结果在精子发生、细胞分化、精子活动和多细胞生物的发育等方面改变更为显著。此外,KEGG结果显示,成熟期精原干细胞在能量代谢方面出现糖酵解作用方式(图5B)。
成熟期精原干细胞与新生期精原干细胞前体细胞相比,有两个显著的生物学功能变化:第一个子网络的富集结果显著集中在与细胞外基质或环境的相互作用关系上;而第二个子网络的富集结果显示成熟期精原干细胞与免疫系统之间存在关联性(图5C),提示小鼠个体成熟后的精原干细胞改变了与细胞外基质的联系方式,如在减少胶原产生、促进胶原分解上表现出明显变化(图5C)。
综合3个发育不同年龄阶段小鼠OCT4阳性精原干细胞聚类子网络的功能富集结果,聚类子网络分析提示成熟期精原干细胞存在产生细胞外泌体、与先天性免疫反应、超敏反应和补体激活有关的特性。例如,能够启动先天免疫反应的TLR3[37,38]在新生期精原干细胞前体细胞中的转录水平最高,幼年期精原干细胞中转录量已显著降低,成熟期精原干细胞中几乎检测不到表达;细胞外泌体标志蛋白CD63[39]在幼年期精原干细胞中未见明显变化,在成熟期精原干细胞中的表达水平有所上调;相反,参与抗体结合的FCGR3和FCER1G,以及主要组织相容性复合体Ⅰ(MHCⅠ)的组成[40]成分H2-K1和H2-D1的转录水平在幼年期下调后又在成熟期显著上调(图6A)。但精原干细胞中补体C1QA、C1QB和C1QC的表达水平在小鼠幼年期未见明显变化,在成熟阶段显著上调(图6B)。先天免疫反应是针对病原体的非特异性免疫反应;而超敏反应是适应性免疫针对特定抗原产生的,以生理功能紊乱为主的异常特异性免疫应答。这些结果提示,幼年期精原干细胞参与先天免疫反应的启动,而成熟期精原干细胞通过产生补体参与先天免疫反应以预防炎症的发生[41],并与超敏反应相关。由于成熟期时有影响巨噬细胞耐受表型[41]的补体(C1QA、C1QB、C1QC)大量表达,从而防止了自身免疫反应的发生。因此,成年小鼠精原干细胞的移植可能存在组织相容性问题。
2.6 HALLMARK基因集富集分析
HALLMARK基因集富集分析可以总结多个基因集的信息,突出显示协调表达和具有明确生物学过程定义的基因,从而减少基因分析的误差和冗余,并为GSEA分析更好地描述生物学功能提供了基础[34]。本研究对不同发育时期精原干细胞中的所有基因进行了HALLMARK基因集富集分析。在新生期精原干细胞前体细胞与幼年期精原干细胞之间的分析结果中,显著性和标准化富集分数排在前5位的生物学功能均呈现下调趋势,分别是上皮间质转化(epithelial-mesenchymal transition, EMT)、IL2_ STAT5信号通路、凝血级联、补体级联和通过NFκB的TNFα信号通路(图7A),该结果提示幼年期精原干细胞EMT、STAT信号和TNFα信号下调,以及凝血级联和补体级联等免疫相关反应下调。在幼年期精原干细胞与成熟期精原干细胞之间的分析结果中,排在前5位的是下调的MYC_TARGETS_V1、氧化磷酸化、上皮间质转化、E2F_TARGETS和上调的精子发生(图7B),说明成熟期精原干细胞EMT持续下调,增殖减慢,氧化磷酸化下调,存在分化趋势。而成熟期精原干细胞与新生期精原干细胞前体细胞之间的分析结果和幼年期与成熟期精原干细胞之间的分析结果相同,但EMT、MYC_TARGETS_ V1和氧化磷酸化的下调程度不同(图7C),该结果表明,与新生期精原干细胞前体细胞相比,成熟期精原干细胞在增殖、EMT和有氧代谢方面显著下调,并出现分化倾向。综上,HALLMARK基因集富集结果表明,小鼠从新生到成熟,OCT4阳性精原干细胞的上皮间质转化是持续下降的,并且出现上调的精子发生过程相关基因表达,说明OCT4阳性精原干细胞在转录水平上存在多能性下降特征,同时出现氧化磷酸化作用逐渐削弱的现象,且成熟精原干细胞的增殖活动减少。
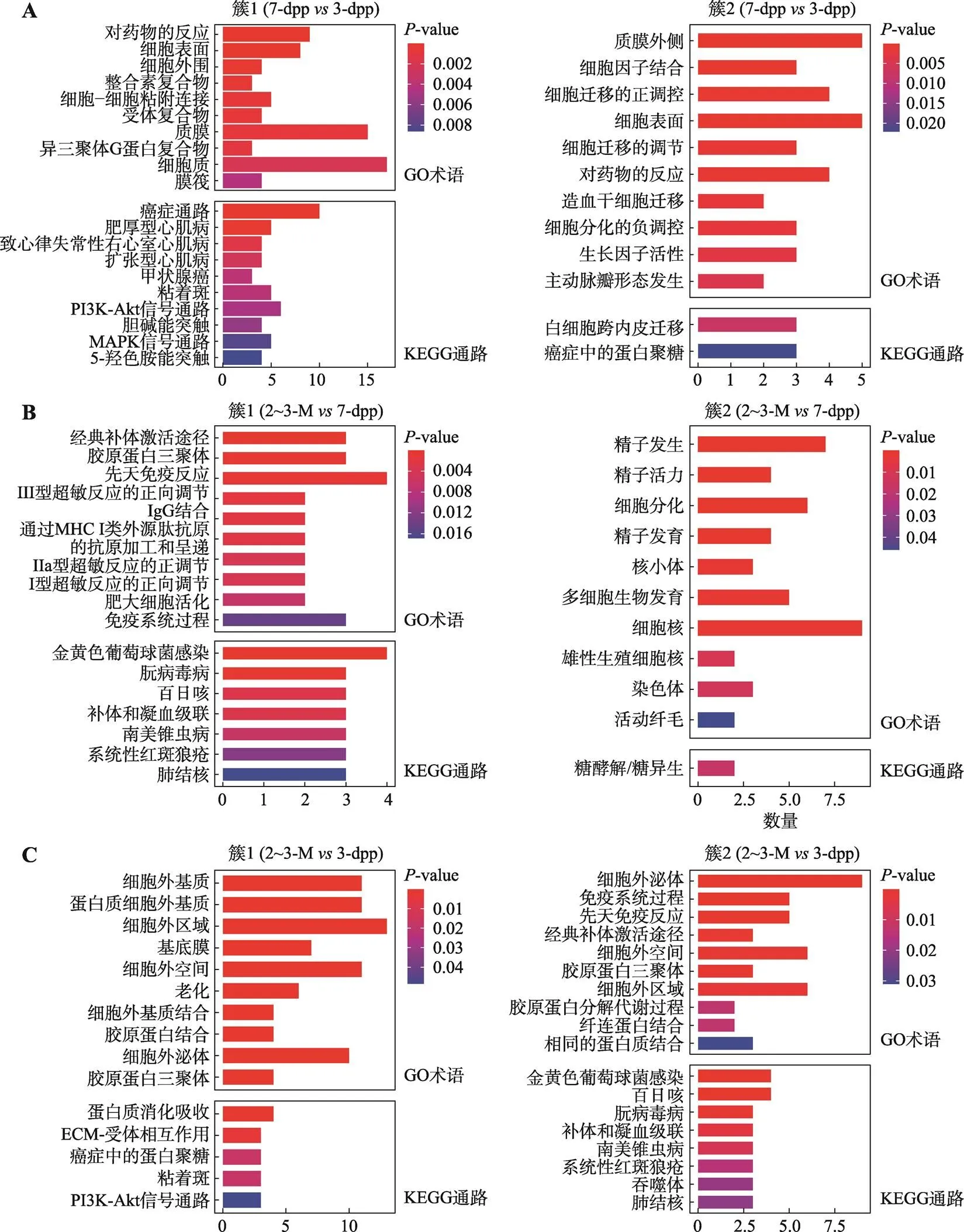
图5 综合评分最高的两个PPI聚类网络簇基因的生物学功能分析
A:7-dpp3-dpp差异基因PPI网络中综合评分最高的两个聚类子网络基因的生物学功能分析结果;B:2~3-M7-dpp差异基因PPI网络中综合评分最高的两个聚类子网络基因的生物学功能分析结果;C:2~3-M3-dpp差异基因PPI网络中综合评分最高的两个聚类子网络基因的生物学功能分析结果。<0.05,x轴为富集基因数,y轴为功能描述。
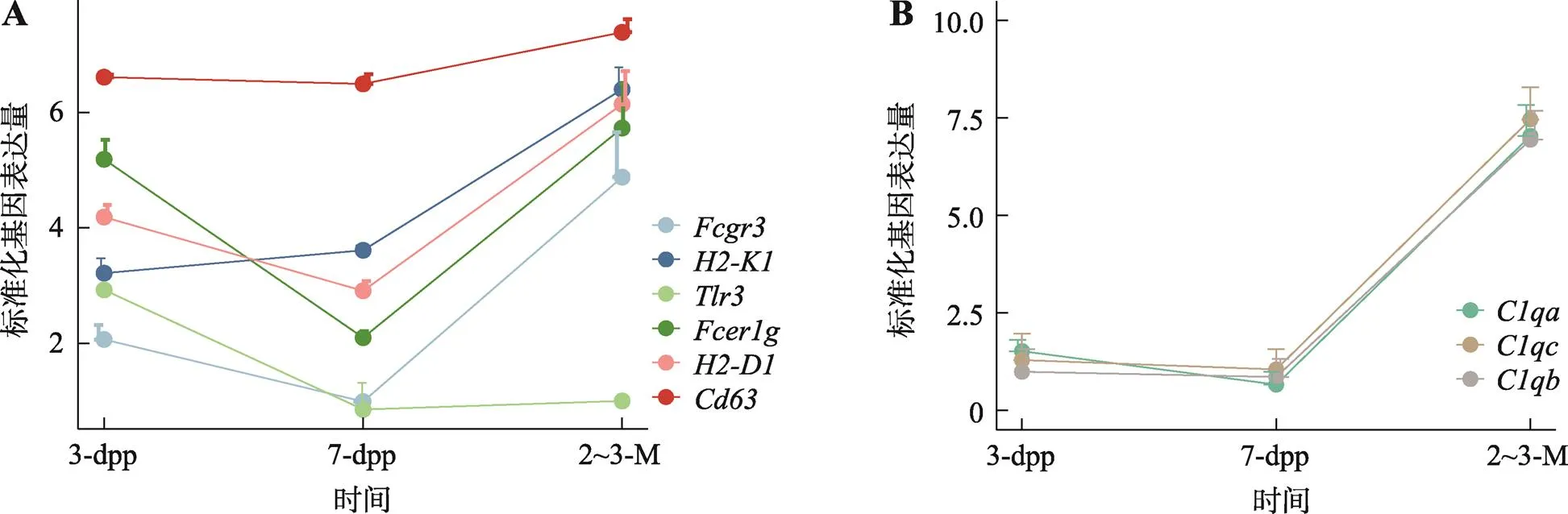
图6 免疫系统相关基因的表达趋势
A:免疫系统相关基因的表达趋势;B:补体基因的表达趋势。数据均以mean±s.e.m表示。

图7 HALLMARK基因集富集分析
A:7-dpp3-dpp样本在HALLMARK基因集富集分析中富集最高的5个条目;B:2~3-M7-dpp样本在HALLMARK基因集富集分析中富集最高的5个条目;C:2~3-M3-dpp样本在HALLMARK基因集富集分析中显著性最高的5个条目。Pathway:功能描述;Gene ranks:基因排序;NES:标准化富集分数;-adj:调整后值。
2.7 潜在信号通路和功能富集分析
为了发现在不同阶段精原干细胞中的潜在信号通路,本研究还采用了基于GO和KEGG基因集的GSEA分析方法。因为GSEA分析方法不局限于用给定的差异基因范围来富集潜在生物学功能,某些在表达上差异不显著,但存在重要生物学意义的基因也会被囊括在富集到的相关潜在信号通路中。该方法不仅可以指出潜在信号通路方向和具有生物学意义的基因,还可以根据整体的基因表达来计算出这些潜在信号通路的上调和下调情况,从而预估潜在信号通路在不同阶段精原干细胞中的生物学意义。
通过基于GO基因集的GSEA分析,新生期精原干细胞前体细胞与幼年期精原干细胞之间共获得潜在GO功能357条,幼年期与成熟期精原干细胞之间的比较共获得潜在GO功能162条,成熟期精原干细胞与新生期精原干细胞前体细胞之间比较共获得潜在GO功能388条,在3个发育阶段OCT4阳性细胞中都具有显著性差异的生物学过程有16个(图8A)。其中13个生物学过程在3个时期的OCT4阳性细胞中持续下调,主要包括:含胶原的细胞外基质、超分子纤维组织、外包裹结构组织、细胞外结构组织、细胞–基质粘附、上皮管分支的形态发生、细胞外基质组装和结构分子活性等,共483个潜在基因;另外3个生物学过程则是持续上调,主要包括:雄性配子生成、精子发生、精细胞分化,共111个潜在基因。对3个上调的生物学过程中的潜在基因经过KEGG通路分析,富集到了多条信号通路,包括:PI3K-Akt、Wnt、Estrogen、Hippo、Rap1、TNF、趋化因子信号通路等,也包含了白细胞跨内皮迁移、紧密连接、间隙连接等相关信号通路(图8B)。13个下调的GO生物学过程的潜在基因经过KEGG通路分析,富集到的信号通路包括:核糖体、粘着斑、细菌侵袭上皮细胞、调节肌动蛋白细胞骨架、ECM-受体相互作用、白细胞跨内皮迁移、癌症中的蛋白多糖、紧密连接、粘附连接以及吞噬体等相关功能(图8C)。通过观察上调和下调的GO生物学功能中潜在基因的转录表达趋势,发现同时出现在两种趋势的GO生物学功能中。紧密连接相关蛋白CLDN11[42]的表达在幼年期精原干细胞中显著上调,但在成熟期精原干细胞中又下调到幼年期转录水平之下(图8D)。这些结果说明,随着小鼠年龄的增长,精原干细胞与细胞外结构组织以及周围细胞间的连接在不断减少,表明精原干细胞与微环境的连接方式发生改变。与此相反,精子发生相关基因的转录表达则随小鼠年龄增加而持续上调。随着小鼠个体的逐渐成熟,OCT4阳性精原干细胞在转录水平上的分化趋势也逐渐明显。
基于KEGG基因集的GSEA分析结果表明,共有8条潜在生物学信号通路在不同年龄小鼠精原干细胞中均具有显著差异(图9A)。如成熟期精原干细胞在免疫反应相关的通路上表现出显著上调,包括抗原加工和呈递、系统性红斑狼疮通路。而物质的合成与线粒体能量代谢在成熟期精原干细胞中有所下降,包括药物代谢–细胞色素P450、谷胱甘肽代谢、药物代谢–其他酶、肌萎缩侧索硬化症、帕金森病和核糖体通路。对富集到的8条潜在生物学通路下的所有基因进行GO分析,结果显示,成熟期精原干细胞在核糖体组装、蛋白质合成、氨基酸代谢、线粒体相关的能量代谢以及线粒体组装等生物合成过程调控显著下调(图9B)。由RNA测序结果可知,随着小鼠年龄的增加,参与线粒体ATP合成偶联质子转运的基因(如、、)、三羧酸循环的基因(如、、)、参与线粒体组织(、、)和线粒体翻译(、、)的基因[43]转录水平在精原干细胞中逐渐下调(图9C)。而参与糖酵解代谢的关键糖酵解酶在成熟期精原干细胞中表达显著上调,如己糖激酶(HK1)、磷酸果糖激酶(PFKFB3)、GAPDHS、烯醇化酶(ENO3)、乳酸脱氢酶(LDHC)、丙酮酸脱氢酶激酶(PDK2)。相反,催化丙酮酸转化为乙酰辅酶A的丙酮酸脱氢酶的表达显著下调,如PDHA1(图9D)。这些结果表明,在小鼠个体发育过程中,OCT4阳性精原干细胞从幼年期开始出现氧化磷酸化作用降低并逐渐提高糖酵解代谢的现象,且伴有核糖体形成减少和整体蛋白质合成水平显著降低的趋势。
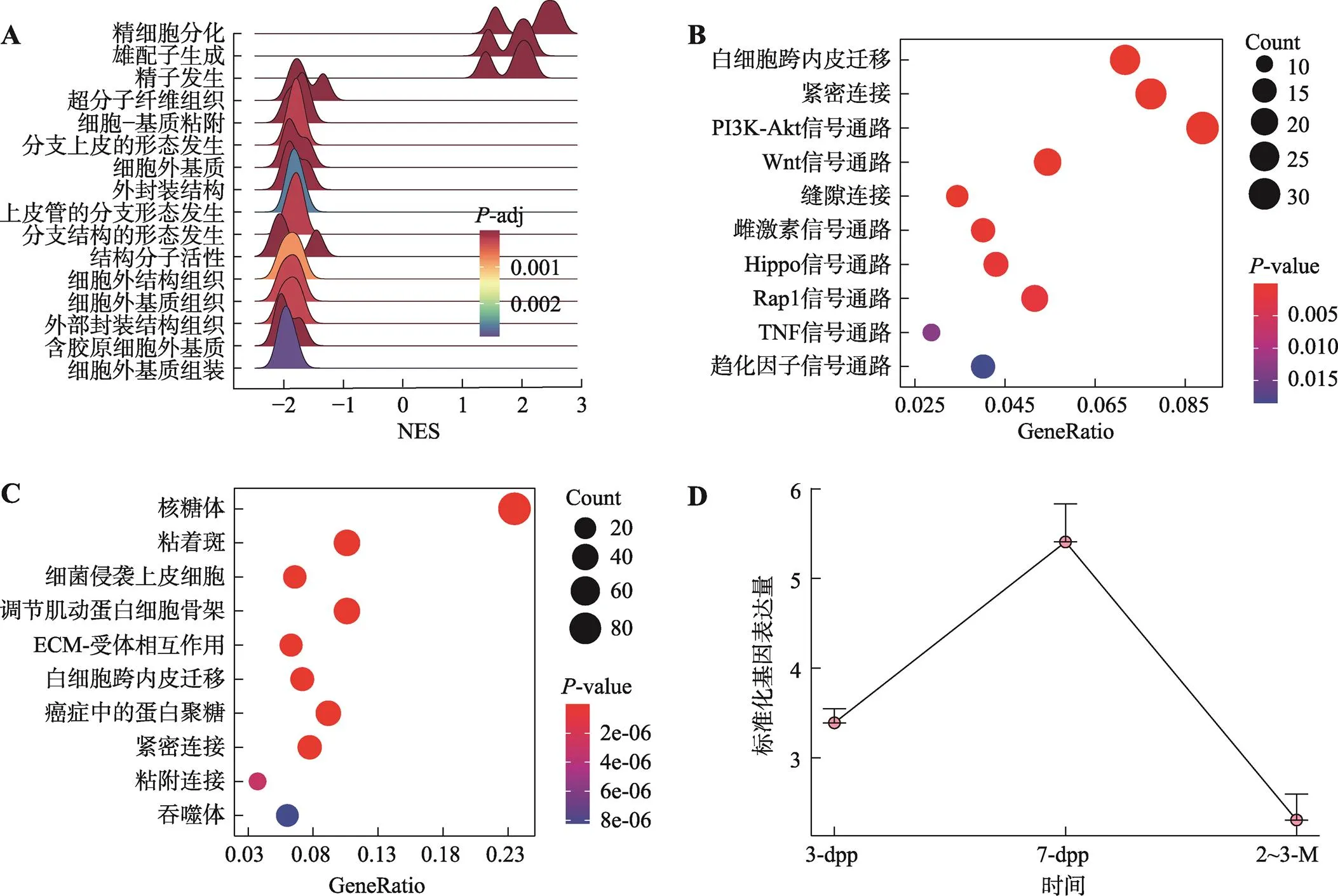
图8 GO基因集富集分析及Cldn11基因表达趋势
A:GSEA分析:持续上调或下调的GO术语。x轴为标准化富集分数,y轴为功能描述。B:上调GO术语基因的KEGG分析(显著性最高的10条通路)。x轴为富集基因占总基因数量的比例,y轴为KEGG通路描述。C:下调GO术语基因的KEGG分析(显著性最高的10条通路)。x轴为富集基因占总基因数量的比例;y轴为KEGG通路描述。颜色表示值范围,点的大小表示通路下富集基因数量。D:基因的表达趋势。数据均以mean±s.e.m表示。
3 讨论
本研究对出生后不同年龄小鼠的OCT4阳性精原干细胞转录组数据进行了生物信息学分析,结果显示,成熟期小鼠精原干细胞与新生期精原干细胞前体细胞及幼年期精原干细胞相比,在细胞膜表面、细胞间或与细胞外基质的粘附、细胞间的连接等方面存在显著性差异,在胶原产生的减少、促进胶原分解上的变化尤为明显。成熟期精原干细胞可以产生细胞外泌体,参与先天性免疫反应、超敏反应和补体激活过程。随着小鼠个体的发育成熟,精原干细胞内的氧化磷酸化作用逐渐减少,糖酵解代谢上调,且伴有整体蛋白质合成能力的显著降低。
精子发生的正常进行需要生殖细胞之间以及生殖细胞与体细胞之间的密切联系和高度配合。精原干细胞在细胞膜上的显著改变值得注意。成熟期精原干细胞中差异基因表达分析结果提示,细胞粘附相关分子和通路在这一时期具有显著变化,这与一些已有的研究报道一致。例如:以猪()精原干细胞为模型的体外实验结果显示,具有三螺旋结构的胶原蛋白IV会强烈地刺激猪精原干细胞的分化[44,45]。相较于新生期和幼年期小鼠,成年小鼠中的精原干细胞表现出显著的胶原三聚体分解,这可能是精原干细胞在形成稳定克隆以后减少了胶原蛋白的产生和连接,从而减少刺激其分化的因素。但精原干细胞表面起连接作用的分子远不止一种。有研究表明,生精小管的支持细胞在成熟期间失去了典型的上皮连接结构,取而代之的是特殊的细胞粘附和连接方式[46]。在小鼠发育成熟过程中,支持细胞分子特征上的这种变化会给睾丸中的生精细胞带来连接分子表达上的变化。例如,CLDN11是紧密连接相关蛋白,基因缺陷造成精原干细胞很难在睾丸中定植和形成克隆[42,47]。相较于幼年期精原干细胞,成熟期精原干细胞中的转录水平显著降低,原因可能是小鼠成熟后睾丸支持细胞改变了细胞间连接方式,从而导致精原干细胞表面粘连蛋白的表达随之改变,包括的表达变化。

图9 基于KEGG基因集的基因集富集分析和线粒体及糖酵解相关基因的表达趋势
A:GSEA分析:持续上调或下调的KEGG通路。x轴为标准化富集分数,y轴为通路描述。B:潜在KEGG通路基因的GO分析(显著性最高的12个生物学功能)。x轴为富集基因占总基因数量的比例;y轴为功能描述。颜色表示值范围,点的大小表示基因数量。C:线粒体相关基因的表达趋势。D:糖酵解相关基因的表达趋势。数据均以mean±s.e.m表示。
睾丸是一个具有免疫特权的器官,虽然免疫系统反应显著降低,但来自血液循环系统和通过男性泌尿生殖道入侵的微生物病原体通常会被清除,这种现象表明睾丸对侵入的病原体具有局部有效的先天免疫反应[48]。已有研究指出,各种模式识别受体在睾丸细胞中大量表达并启动睾丸先天免疫反应。小鼠出生后7天的精原细胞和21天的精母细胞中的TLR3以及精细胞中的TLR11能够启动先天免疫反应[37,38]。RNA测序结果显示,TLR3在新生期精原干细胞前体细胞中的表达量最高,在幼年期精原干细胞中表达量显著降低,在成熟期精原干细胞中几乎检测不到表达,但补体C1QA、C1QB和C1QC在成熟期精原干细胞中的表达水平显著上调。产生这些现象的一种可能性是OCT4阳性精原干细胞在小鼠出生后或许承担着一定的启动先天免疫反应的作用,随着睾丸组织的成熟,陆续形成的精母细胞和精细胞开始负责启动先天免疫反应,而精原干细胞主要通过分泌补体参与其中。
具有免疫特权的睾丸组织除了具有局部细胞先天免疫防御功能,还可以保护自身抗原性生殖细胞免受免疫系统的攻击。RNA测序数据分析富集到的通路中有多条与自身免疫相关,例如抗原加工和呈递、系统性红斑狼疮以及3种超敏反应等,说明除了被血睾屏障保护的减数分裂后生殖细胞具有细胞表面抗原外,精原干细胞可能也具有免疫原性,这与已报道的血睾屏障之外的生殖细胞表达自身抗原具有一致性[49,50]。本研究也从OCT4阳性精原干细胞转录组数据中发现了主要组织相容性复合体分子的转录表达随年龄的增加而上调。本研究发现,成熟期精原干细胞具有分泌补体的能力,而补体的作用不局限于协助先天性和适应性免疫反应,同时也可以调节自身免疫耐受[41]。因此,OCT4阳性精原干细胞分泌的补体可能也为避免自身免疫的发生提供了一定的帮助。这些结果说明精原干细胞在睾丸免疫特权中可能发挥着一定的作用,并且成体精原干细胞的非自体移植可能引起自身免疫性反应。
目前普遍认为,多种成体干细胞均具有低线粒体呼吸和无氧代谢特征,线粒体氧化磷酸化作用持续产生的潜在有害活性氧减少,从而维护基因组的完整性[51]。代谢方式的转变似乎是生命体成长发育和命运决定所必须的,并存在于多个组织中[52]。例如,植入前胚胎被认为依赖丙酮酸消耗,偏爱高度氧化代谢[53~55],而在囊胚形成时糖酵解急剧增加[56~58]。原始生殖细胞被认为依赖线粒体呼吸,而伴随氧化磷酸化产生的中间代谢产物是表观遗传变化所必需的[59~61]。那么,从原始生殖细胞到新生期精原干细胞前体细胞,到幼年期精原干细胞,直至成熟期精原干细胞,代谢方式的转变可能是生殖细胞发育的必然过程。在猪中,二月龄动物精原干细胞中可检测到代谢方式从氧化磷酸化开始转变为厌氧代谢[62]。已报道的睾丸细胞转录组学分析也表明,未分化精原细胞中糖酵解相关基因大量富集以及分化过程中氧化磷酸化相关基因的表达上调[43,63]。小鼠3个发育时间点的OCT4阳性精原干细胞的RNA测序分析结果显示,幼年期精原干细胞尚未出现糖酵解代谢或氧化磷酸化的下降,但在成熟期精原干细胞中出现了显著的氧化磷酸化降低和糖酵解的上调。因此,糖酵解代谢或许不是精原干细胞建立时的主要代谢途径,而随发育和细胞内部分子网络的变化调整,才形成为有利于精原干细胞增殖和长期维持干性的代谢方式,这可能也是生殖细胞从发育潜能性高且依赖有氧代谢的原始生殖细胞阶段,逐渐转变成高分化特性和依赖糖酵解代谢的成熟期精原干细胞的细胞命运变化所必经的过程。然而,本研究存在一定的局限性,因为细胞中发挥实际作用的大部分是蛋白质,需要与精原干细胞蛋白质组学结合,以进一步验证转录组分析的结果。
综上所述,成熟期精原干细胞与新生期精原干细胞前体细胞以及幼年期精原干细胞之间,存在多方面的显著基因表达差异。这些基因表达的改变可以在伴随小鼠个体成熟变化的同时,更好地维护精原干细胞的自我更新,保证周期性精子发生的有序进行,并让后代拥有完整的基因组。虽然精原干细胞还没有很好的特异性单一标记物,但OCT4阳性精原干细胞亚群的转录组研究和分析,可以为精原干细胞特性的了解和研究提供一定的参考。
附加材料详见文章电子版www.chinagene.cn。
附表1 qRT-PCR引物序列
Supplementary Table 1 Primer sequences for qRT-PCR
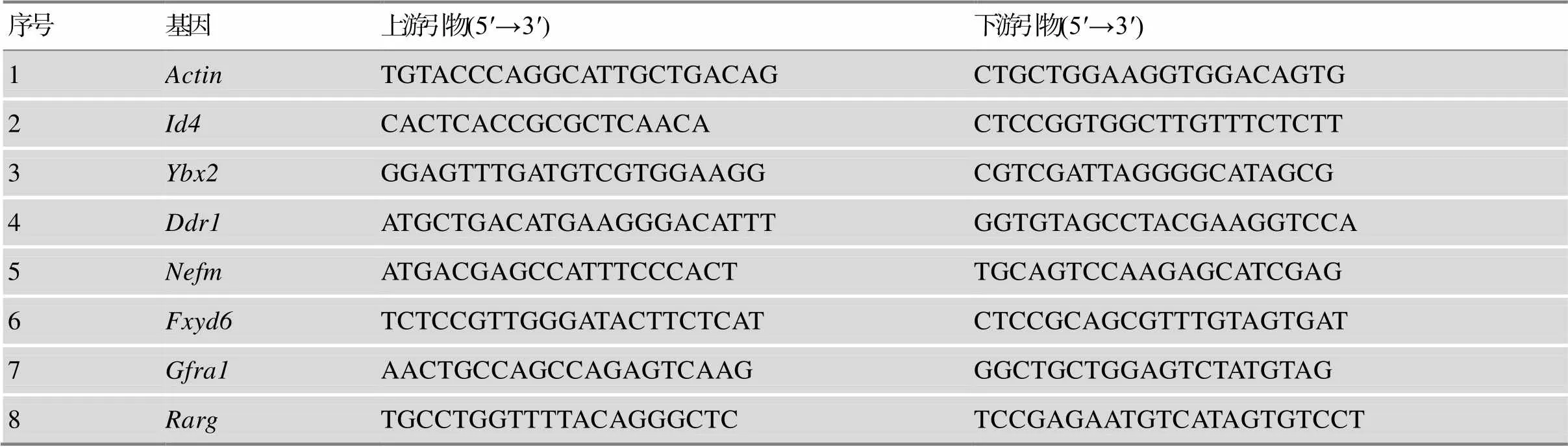
序号基因上游引物(5′→3′)下游引物(5′→3′) 1ActinTGTACCCAGGCATTGCTGACAGCTGCTGGAAGGTGGACAGTG 2Id4CACTCACCGCGCTCAACACTCCGGTGGCTTGTTTCTCTT 3Ybx2GGAGTTTGATGTCGTGGAAGGCGTCGATTAGGGGCATAGCG 4Ddr1ATGCTGACATGAAGGGACATTTGGTGTAGCCTACGAAGGTCCA 5NefmATGACGAGCCATTTCCCACTTGCAGTCCAAGAGCATCGAG 6Fxyd6TCTCCGTTGGGATACTTCTCATCTCCGCAGCGTTTGTAGTGAT 7Gfra1AACTGCCAGCCAGAGTCAAGGGCTGCTGGAGTCTATGTAG 8RargTGCCTGGTTTTACAGGGCTCTCCGAGAATGTCATAGTGTCCT
[1] Vander Borght M, Wyns C. Fertility and infertility: definition and epidemiology., 2018, 62: 2–10.
[2] Agarwal A, Baskaran S, Parekh N, Cho CL, Henkel R, Vij S, Arafa M, Panner Selvam MK, Shah R. Male infertility., 2021, 397(10271): 319–333.
[3] Phillips BT, Gassei K, Orwig KE. Spermatogonial stem cell regulation and spermatogenesis., 2010, 365(1546): 1663–1678.
[4] Mei XX, Wang J, Wu J. Extrinsic and intrinsic factors controlling spermatogonial stem cell self-renewal and differentiation., 2015, 17(3): 347–354.
[5] Jan SZ, Vormer TL, Jongejan A, Röling MD, Silber SJ, de Rooij DG, Hamer G, Repping S, van Pelt AMM. Unraveling transcriptome dynamics in human spermatogenesis., 2017, 144(20): 3659–3673.
[6] Izadyar F, Den Ouden K, Stout TAE, Stout J, Coret J, Lankveld DPK, Spoormakers TJP, Colenbrander B, Oldenbroek JK, Van der Ploeg KD, Woelders H, Kal HB, De Rooij DG. Autologous and homologous transplantation of bovine spermatogonial stem cells., 2003, 126(6): 765–774.
[7] Schlatt S, Foppiani L, Rolf C, Weinbauer GF, Nieschlag E. Germ cell transplantation into X-irradiated monkey testes., 2002, 17(1): 55–62.
[8] Jahnukainen K, Ehmcke J, Quader MA, Saiful Huq M, Epperly MW, Hergenrother S, Nurmio M, Schlatt S. Testicular recovery after irradiation differs in prepubertal and pubertal non-human primates, and can be enhanced by autologous germ cell transplantation., 2011, 26(8): 1945–1954.
[9] Hermann BP, Sukhwani M, Winkler F, Pascarella JN, Peters KA, Sheng Y, Valli H, Rodriguez M, Ezzelarab M, Dargo G, Peterson K, Masterson K, Ramsey C, Ward T, Lienesch M, Volk A, Cooper DK, Thomson AW, Kiss JE, Penedo MCT, Schatten GP, Mitalipov S, Orwig KE. Spermatogonial stem cell transplantation into rhesus testes regenerates spermatogenesis producing functional sperm., 2012, 11(5): 715–726.
[10] Nakagawa T, Sharma M, Nabeshima Y, Braun RE, Yoshida S. Functional hierarchy and reversibility within the murine spermatogenic stem cell compartment., 2010, 328(5974): 62–67.
[11] Carrieri C, Comazzetto S, Grover A, Morgan M, Buness A, Nerlov C, O'Carroll D. A transit-amplifying population underpins the efficient regenerative capacity of the testis., 2017, 214(6): 1631–1641.
[12] La HM, Mäkelä JA, Chan AL, Rossello FJ, Nefzger CM, Legrand JMD, De Seram M, Polo JM, Hobbs RM. Identification of dynamic undifferentiated cell states within the male germline., 2018, 9(1): 2819.
[13] Clevers H, Watt FM. Defining adult stem cells by function, not by phenotype., 2018, 87: 1015– 1027.
[14] Mäkelä JA, Hobbs RM. Molecular regulation of spermatogonial stem cell renewal and differentiation., 2019, 158(5): R169–R187.
[15] Nagano M, Avarbock MR, Brinster RL. Pattern and kinetics of mouse donor spermatogonial stem cell colonization in recipient testes., 1999, 60(6): 1429–1436.
[16] Nagano MC. Homing efficiency and proliferation kinetics of male germ line stem cells following transplantation in mice., 2003, 69(2): 701–707.
[17] Forbes CM, Flannigan R, Schlegel PN. Spermatogonial stem cell transplantation and male infertility: current status and future directions., 2017, 16(1): 171–180.
[18] Schmidt JA, Abramowitz LK, Kubota H, Wu X, Niu Z, Avarbock MR, Tobias JW, Bartolomei MS, Brinster RL.andaging is detrimental to mouse spermatogonial stem cell function., 2011, 84(4): 698–706.
[19] Kanatsu-Shinohara M, Ogonuki N, Iwano T, Lee J, Kazuki Y, Inoue K, Miki H, Takehashi M, Toyokuni S, Shinkai Y, Oshimura M, Ishino F, Ogura A, Shinohara T. Genetic and epigenetic properties of mouse male germline stem cells during long-term culture., 2005, 132(18): 4155–4163.
[20] Subash SK, Kumar PG. Spermatogonial stem cells: a story of self-renewal and differentiation., 2021, 26: 163–205.
[21] Zhao X, Yang HQ. Progress on spermatogonial stem cells of large animals., 2019, 41(8): 686– 702.赵鑫, 杨化强. 大动物精原干细胞研究进展. 遗传, 2019, 41(8): 686–702.
[22] Mäkelä JA, Toppari J. Spermatogenesis. In: Simoni M, Huhtaniemi I, eds. Endocrinology of the Testis and Male Reproduction. Endocrinology. Springer Cham, 2017, 417–455.
[23] Liao JY, Suen HC, Luk ACS, Yang LL, Lee AWT, Qi HY, Lee TL. Transcriptomic and epigenomic profiling of young and aged spermatogonial stem cells reveals molecular targets regulating differentiation., 2021, 17(7): e1009369.
[24] Yang LL, Wu W, Qi HY. Gene expression profiling revealed specific spermatogonial stem cell genes in mouse., 2013, 51(2): 83–96.
[25] Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements., 2015, 12(4): 357–360.
[26] Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., 2014, 15(12): 550.
[27] Jiang YA, Leng JX, Lin QX, Zhou F. Epithelial- mesenchymal transition related genes in unruptured aneurysms identified through weighted gene coexpression network analysis., 2022, 12(1): 225.
[28] Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets., 2021, 49(D1): D605–D612.
[29] Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks., 2003, 13(11): 2498–2504.
[30] Bader GD, Hogue CWV. An automated method for finding molecular complexes in large protein interaction networks., 2003, 4: 2.
[31] Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery., 2003, 4(5): P3.
[32] Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0., 2011, 27(12): 1739–1740.
[33] Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles., 2005, 102(43): 15545–15550.
[34] Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection., 2015, 1(6): 417–425.
[35] Liberzon A. A description of the Molecular Signatures Database (MSigDB) Web site., 2014, 1150: 153–160.
[36] Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing., 1995, 57(1): 289–300.
[37] Wang T, Zhang XY, Chen QY, Deng TT, Zhang Y, Li N, Shang T, Chen YM, Han DS. Toll-like receptor 3-initiated antiviral responses in mouse male germ cells., 2012, 86(4): 106.
[38] Chen QY, Zhu WW, Liu ZH, Yan KQ, Zhao ST, Han DS. Toll-like receptor 11-initiated innate immune response in male mouse germ cells., 2014, 90(2): 38.
[39] Mathieu M, Névo N, Jouve M, Valenzuela JI, Maurin M, Verweij FJ, Palmulli R, Lankar D, Dingli F, Loew D, Rubinstein E, Boncompain G, Perez F, Théry C. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9., 2021, 12(1): 4389.
[40] Mangold CA, Masser DR, Stanford DR, Bixler GV, Pisupati A, Giles CB, Wren JD, Ford MM, Sonntag WE, Freeman WM. CNS-wide sexually dimorphic induction of the major histocompatibility Complex 1 pathway with aging., 2017, 72(1): 16–29.
[41] Son M, Diamond B, Santiago-Schwarz F. Fundamental role of C1q in autoimmunity and inflammation., 2015, 63(1–3): 101–106.
[42] Kanatsu-Shinohara M, Ogonuki N, Matoba S, Ogura A, Shinohara T. Autologous transplantation of spermatogonial stem cells restores fertility in congenitally infertile mice., 2020, 117(14): 7837–7844.
[43] Lord T, Nixon B. Metabolic changes accompanying spermatogonial stem cell differentiation., 2020, 52(4): 399–411.
[44] Park MH, Park JE, Kim MS, Lee KY, Hwang JY, Yun JI, Choi JH, Lee E, Lee ST. Effects of extracellular matrix protein-derived signaling on the maintenance of the undifferentiated state of spermatogonial stem cells from porcine neonatal testis., 2016, 29(10): 1398–1406.
[45] Shinohara T, Avarbock MR, Brinster RL. Beta1- and alpha6-integrin are surface markers on mouse spermatogonial stem cells., 1999, 96(10): 5504–5509.
[46] Domke LM, Rickelt S, Dörflinger Y, Kuhn C, Winter-Simanowski S, Zimbelmann R, Rosin-Arbesfeld R, Heid H, Franke WW. The cell-cell junctions of mammalian testes: I. The adhering junctions of the seminiferous epithelium represent special differentiation structures., 2014, 357(3): 645–665.
[47] Morimoto H, Ogonuki N, Kanatsu-Shinohara M, Matoba S, Ogura A, Shinohara T. Spermatogonial stem cell transplantation into nonablated mouse recipient testes., 2021, 16(7): 1832–1844.
[48] Zhao ST, Zhu WW, Xue SP, Han DS. Testicular defense systems: immune privilege and innate immunity., 2014, 11(5): 428–437.
[49] Yule TD, Montoya GD, Russell LD, Williams TM, Tung KS. Autoantigenic germ cells exist outside the blood testis barrier., 1988, 141(4): 1161–1167.
[50] Setchell BP. The testis and tissue transplantation: historical aspects., 1990, 18(1): 1–8.
[51] Formosa LE, Ryan MT. Mitochondrial OXPHOS complex assembly lines., 2018, 20(5): 511–513.
[52] Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, Bomze D, Elena-Herrmann B, Scherf T, Nissim-Rafinia M, Kempa S, Itskovitz-Eldor J, Meshorer E, Aberdam D, Nahmias Y. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells., 2015, 21(3): 392–402.
[53] Voigt AL, Thiageswaran S, de Lima E Martins Lara N, Dobrinski I. Metabolic requirements for spermatogonial stem cell establishment and maintenanceand., 2021, 22(4): 1998.
[54] Brinster RL, Troike DE. Requirements for blastocyst development., 1979, 49 Suppl 2: 26–34.
[55] Butcher L, Coates A, Martin KL, Rutherford AJ, Leese HJ. Metabolism of pyruvate by the early human embryo., 1998, 58(4): 1054–1056.
[56] Gardner DK, Lane M, Stevens J, Schoolcraft WB. Noninvasive assessment of human embryo nutrient consumption as a measure of developmental potential., 2001, 76(6): 1175–1180.
[57] Leese HJ, Barton AM. Pyruvate and glucose uptake by mouse ova and preimplantation embryos., 1984, 72(1): 9–13.
[58] Leese HJ. Metabolism of the preimplantation embryo: 40 years on., 2012, 143(4): 417–427.
[59] Tischler J, Gruhn WH, Reid J, Allgeyer E, Buettner F, Marr C, Theis F, Simons BD, Wernisch L, Surani MA. Metabolic regulation of pluripotency and germ cell fate through α-ketoglutarate., 2019, 38(1): e99518.
[60] Yoshida S. Open niche regulation of mouse spermatogenic stem cells., 2018, 60(9): 542–552.
[61] Hayashi Y, Otsuka K, Ebina M, Igarashi K, Takehara A, Matsumoto M, Kanai A, Igarashi K, Soga T, Matsui Y. Distinct requirements for energy metabolism in mouse primordial germ cells and their reprogramming to embryonic germ cells., 2017, 114(31): 8289–8294.
[62] Voigt AL, Kondro DA, Powell D, Valli-Pulaski H, Ungrin M, Stukenborg JB, Klein C, Lewis IA, Orwig KE, Dobrinski I. Unique metabolic phenotype and its transition during maturation of juvenile male germ cells., 2021, 35(5): e21513.
[63] Sohni A, Tan K, Song HW, Burow D, de Rooij DG, Laurent L, Hsieh TC, Rabah R, Hammoud SS, Vicini E, Wilkinson MF. The neonatal and adult human testis defined at the single-cell level., 2019, 26(6): 1501–1517.e4.
Transcriptome analysis of mouse male germline stem cells reveals characteristics of mature spermatogonial stem cells
Yan Guo1, Lele Yang2, Huayu Qi2
Spermatogonial stem cells (SSCs) are adult stem cells in the testis of male animals and have the ability in self-renewal and differentiation. SSCs are derived from primordial germ cells (PGCs) that are mitotically arrested in the embryo before birth. Following the birth of the animal, PGCs resume mitosis and migrate from the centre of the seminiferous tubules to the basement membrane. The descendent of PGCs (also called gonocytes) establish stable SSC colonies in about a week postnatally in order to support the life-long spermatogenesis. Whether SSCs at different developmental stages differ in their molecular and cellular characteristics is currently unclear. In the presented study, we conducted bioinformatics analyses using transcriptomics data established previously in the laboratory on OCT4 (encoded by the pluripotent gene) expressing SSCs from the neonatal (3 days-post-partum, 3-dpp), juvenile (7-dpp) and adult (2~3-month) mice, including screen of differentially expressed genes (DEGs), protein-protein interaction (PPI) network analysis of DEGs and clustering of sub-networks from PPI. GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) analyses were also performed on clustered sub-networks of the PPI. In addition, all genes were analyzed using GSEA (gene set enrichment analysis) based on GO, KEGG and HALLMARK gene sets. The results showed that SSCs have a large number of DEGs among OCT4-positive SSCs from neonatal, juvenile and adult mice. The distinguishable biological functions encoded by these DEGs include biosynthesis and energy metabolism, immune response, cell junction and expression of migration and cell differentiation-related genes. Significant changes in the cell membrane composition of OCT4-positive SSCs may not only cause hypersensitive immune reactions but also affect the cell-cell contact and responses to secreted cytokines in the extracellular environment. The results also suggest that OCT4-positive SSCs may shift metabolic state from oxidative phosphorylation to glycolysis and significantly reduce the transcription of genes related to ribosome formation during aging. These results provide new clues for future research on the regulatory mechanisms of male germline stem cell development, growth and aging.
spermatogonial stem cells; OCT4; transcriptome; differentially expressed genes
(责任编委: 刘默芳)
