Formation and Collapse of Cellulose Nanocrystals and Hydrophobic Association-induced Dual Cross-linked Nanocomposite Hydrogels:A Rheological Study
2022-07-21XiaoruiJinMingguoMaJunYang
Xiaorui Jin,Mingguo Ma,Jun Yang
Beijing Key Laboratory of Lignocellulosic Chemistry,College of Materials Science and Technology,Beijing Forestry University,Beijing,100083,China
Abstract: The rheological characteristics of a physical gelation system, in which cellulose nanocrystals (CNCs) induced the entanglement of poly(acrylic acid)(PAA)chains and partial hydrophobic association of octylphenol polyoxyethylene acrylate (OP-10-AC) branches in a micellar solution of sodium dodecyl sulfate (SDS), were investigated.The gelation time of the physical gels decreased as the CNC content and number of hydrophobic branch units increased.At the gel point, the storage modulus (G') and loss modulus (G") followed the same frequency dependence (G' ≈G" ≈ωn),where the hydrophobic moieties attached to the side chains had a significant impact on the values of viscoelastic exponent (n).Beyond the gel point, the initial polymer solution was transformed to a solid-like gel, and the strength of the gel network was governed by associations between both the CNCs and hydrophobic groups.The evolution of the viscoelasticity during the gel-sol transition was monitored, demonstrating that due to a reversible arrangement of the hydrophobic units, a large proportion of physical cross-links dissociated under a thermal trigger and were reversibly reformed when the solution was cooled, while no such partial recovery was observed in the case of the single CNC-induced network systems(with no hydrophobic branches).
Keywords: hydrogels; cellulose nanocrystals; hydrophobic association;rheology
1 lntroduction
Polymeric hydrogels consisting of three-dimensional cross-linked networks are a most interesting class of"soft matter" with several established and many more potential applications as hydrophilic functional materials.Polymeric hydrogels are used as water absorbents and tissue engineering scaffolds, as well as in enzyme catalysis and membrane processes[1-2].Hydrogels possess the ability to absorb water many times their own weight without dissolving owing to the presence of chemical or physical cross-links.In chemically cross-linked hydrogels, the polymer chains are interconnected by permanent covalent bonds,whereas in physically cross-linked hydrogels, the polymer chains generally aggregate via non-covalent bonds such as van der Waals force, hydrogen bonding,or hydrophobic associations[3].
To bridge customized and synthetic chemical networks to achieve reversibility and flexibility of the physical network, supermolecular polymer networks have recently been developed as a promising new class of materials[4].These networks are generally formed by hydrogen bonding, hydrophobic associations, or π-π interactions, which serve to form networks by noncovalent association of monomers or chains.This type of polymer network possesses both chemical and physical advantages.The polymers can be custommade and designed for targeted applications,presenting high strength under favorable conditions,and are easily de-crosslinked or de-associated by tailoring the system conditions[5].A copolymer incorporating both soluble fragments and a small fraction of insoluble components that strongly attach to each other is normally called an associative polymer[6-7].Different chemical units can be viewed as associations, depending on the specific solvents (such as hydrophobic alkyl fragments for aqueous solutions and polar or ionic groups for organic solvents).The number of associations per aggregation depends on the size of the association and the attraction energy.Moreover,the formation of transient associations between macromolecules can provide temporary junction domains, resulting in significant modifications of the polymer properties, such as thickening, shearthinning, or gelation.For example, in aqueous systems,the formation of reversible gels by hydrophobicmodified polymers is regarded as a self-association process, and gels can be obtained by incorporating molecules or particles that reversibly bridge crosslinked networks[8].Similarly,incorporating hydrophobic associations into solutions with additives or particles(e.g., surfactants, vesicles, nanometer sheets, or lipid lamellae) to strengthen the hydrophobic associations between polymers has also been widely reported[6-8].Thus, due to this inter-attraction, the hydrophobic associations can aggregate and form multiple morphologies above a certain polymer concentration,where interconnected polymer chains form gel-like clusters (physical gels) and play a role analogous to cross-links[9-10].
Recently, we utilized cellulose nanocrystals (CNCs)as both organic fillers and anchors to graft hydrophilic polymers (e.g., poly(acrylic acid) (PAA)[11]and poly(acrylamide) (PAM)[12]).The obtained hybrid gels exhibited greater tensile strength, Young's modulus,and elongation at break than their conventional chemically cross-linked counterparts.This extraordinary mechanical behavior contributed to the formation of a rearrangeable network structure with an efficient energy-dissipation mechanism[12].Moreover, partially hydrophobically modified dual-network gels were synthesized by copolymerization of the hydrophilic monomer acrylic acid (AA) in the presence of a small fraction of the hydrophobic co-monomer octylphenol polyoxyethylene acrylate (OP-10-AC) via micellar polymerization.Owing to the high local concentration of the hydrophobic association micro-domains(HAMDs) within the micelles were distributed as random blocks along the hydrophilic polymer backbones.Thus, this combination of "hard" (CNCs)and "soft" (HAMDs) building blocks afforded unique dual cross-linked nanocomposite hydrogels with higher strength than the single-CNC induced gels: the CNCs and HAMDs were expected to reflect the generic properties of mixtures containing "hard" and "soft"components, respectively, offering opportunities for constructing a physical gel with tunable properties, in the absence of an organic cross-linking agent(Fig.1).
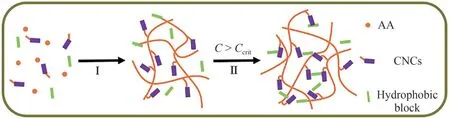
Fig.1 Schematic of transition from dilute solution to cross-linked networks by surface in situ "graft-from" polymerization of CNCs,involving two stages(I and II)
It can be seen that initially hydrophilic monomers(AA), hydrophobic blocks (OP-10-AC), and nanoparticles (CNCs) are well dispersed in dilute solutions before initiation.The monomers graftedin situon the CNC surface form multi-arm-like freemoving macromolecular precursors, where the hydrophobic blocks are randomly distributed along the hydrophilic backbones that solubilize within micelles.These sticky particles are susceptible to association via cluster aggregation.When the particle concentration exceeds a critical value(C>Ccrit),the microgel particles are able to aggregate, where the initial liquid-like small clusters become increasingly interconnected until the rheological gel point and form percolating physical gel networks.
The properties of amphiphilic copolymers containing both hydrophilic and hydrophobic blocks can be easily tailored, enabling extensive applications.It has been reported that a transient network stems from the selfassembly of associating polymers, where the polymer chains can build an aggregated network via hydrophobic interactions.Despite intensive efforts to explore the critical gelation conditions and mechanical properties of CNC/PAA hydrophobic-modified hydrogels,comprehensive rheological characterization and understanding of the detailed evolution of the gelation process remain lacking.To address these issues, this study seeks to gain deeper insight into the factors governing the evolution of viscoelasticity during the gelation process.It is known that rubberylike materials formed via hydrophobic associations may exhibit a reversible gel-sol transition (break and reform dynamically before fracture of the molecular backbone)[6-7].Thus, another topic of this study is to determine whether the incorporation of hydrophobic blocks into the PAA backbone would produce a similar recovery.As demonstrated herein, owing to the hydrophobic interactions that are sensitive to environmental variation, by cooling the solution after the thermally induced chain disentanglement, the dynamic nature of the hydrophobic interactions among polymer chains leads to partial network recovery (gelsol reversible transition), which is a distinctive feature of the as-prepared gel systems.
2 Experimental
2.1 Materials
Acryloyl chloride (AC), AA, sodium dodecyl sulfate(SDS), and octylphenol polyoxyethylene ether (OP-10,where 10 is the number of ethoxy units in each molecule,Mw=647)were used as received.Deionized water was used for all experiments, and the oxygen present in water was removed by bubbling with N2gas for 30 min prior to use.A suspension of CNCs(~1.0 wt%) with an average length of (200±10) nm and width of (30±5) nm was obtained from Tianjin University of Science and Technology.The surface of the CNCs was chemically modified with vinyl groups through treatment withγ-methacryloxypropyl trimethoxy silane; the vinyl groups were used to anchor the polymer chains[11].OP-10-AC was prepared by the reaction of OP-10 acrylate with AC[13].Subsequently,the hydrophobic OP-10-AC was used for micellar copolymerization of AA in the presence of ammonium persulfate ((NH4)2S2O8) as the initiator.The gel preparation procedures were the same as in our earlier work and are briefly described as follows[11]: an aqueous solution containing predetermined water,CNCs, AA, SDS, and OP-10-AC was prepared and dispersed by ultrasonication for 15 min at 300 W.Free radical polymerization was initiated by adding(NH4)2S2O8solution and reacting under N2atmosphere at 50℃ for 2 h.The total monomer (AA+OP-10-AC)concentration was fixed at 10 wt%; the detailed compositions are listed in Table 1.To remove any water-soluble species, the as-prepared hydrogels were immersed in excess water and thoroughly dialyzed for four days.To prepare linear PAA, a certain amount of AA solution was prepared, and free radical polymerization was triggered by (NH4)2S2O8in the absence of CNCs and hydrophobic fractions.

Table 1 Compositions for hydrogel preparation
2.2 Rheology
Oscillatory shear rheology measurements were conducted using a AR2000 rheometer (TA, USA)operating with cone geometry under N2atmosphere.The instrument was equipped with a Peltier device for temperature control.To minimize dehydration, the sample surface was covered with a thin layer of lowviscosity silicone oil.The storage modulus (G') and loss modulus (G") were recorded by subjecting the samples to a dynamic oscillatory test.From the strain sweep oscillatory tests, the linear viscoelastic region was determined to extend up to a strain of 0.05.Thus,the frequency sweep measurement was performed over the range of 0.01-100 Hz at a strain of 0.01.To monitor the gel-sol reversible transition and the hydrophobic interaction-induced hydrogel recovery behavior, theG' values of the swollen gels were determined by heating the gels from 25℃ to 75℃ at a rate of 0.5℃/min, followed by cooling to 25℃ at a rate of 0.5℃/min.
2.3 Uniaxial elongation measurements
The mechanical properties of the hydrogels were tested using an electronic universal material-testing machine(Z005 Materials Tester, Zwick, Germany).The measurement conditions were as follows: crosshead speed of 30 mm/min, gauge length of 20 mm, and hydrogel size of 5 mm height × 40 mm length × 8 mm width.Raw data were recorded as "force versus displacement" and converted to "stress versus strain"with respect to the initial sample dimensions (5 mm ×8 mm).For each group of tests, three parallel samples were analyzed to ensure reproducibility of the stressstrain curves.
2.4 TEM observation
Microscopic observations were performed via transmission electron microscopy (TEM) using a JEM-1010 (JEOL, Japan) instrument at an acceleration voltage of 80 kV.Thin samples were prepared using a microtome, ultrasonicated for 10 min at 300 W, and placed on copper grids for TEM observation.
3 Results and discussion
3.1 Rheological properties
Before discussing the rheological changes during the gelation process, it is worth spending some time discussing the viscosity properties of the linear polymer chains at 25℃(in the absence of CNCs and at HAMD concentrations far below the critical concentration that could form a cross-linked network structure).The flow curves of the solution viscosity as a function of the shear rate for different fractions of the hydrophobic block are illustrated in Fig.2.Notably,the viscosity (|η*(ω)|) of the linear PAA solution was low,virtually Newtonian fluid features were observed at a low shear rate, and non-Newtonian behavior appeared at a high shear rate.For the samples with a small fraction of hydrophobic branches on the backbone(HAMD 0.2% and HAMD 0.6%), the hydrophobic modified PAA polymer chains also exhibited a shear thinning phenomenon, and this trend became more pronounced than in the case of the linear PAA chains.This shear thinning effect was evident when the concentration of hydrophobic branches was higher.The observations indicate that(1)the polymer chains with a branched structure lead to more pronounced entanglement, and (2) the hydrophobic moieties reside inward relative to the backbone at low HAMD concentrations (0.2 wt%), whereas a lightly entangled conformation was produced by intra-molecular chain association at a higher concentration (0.6 wt%).In addition, the viscosity increased with an increase in the concentration of hydrophobic associations in the system.
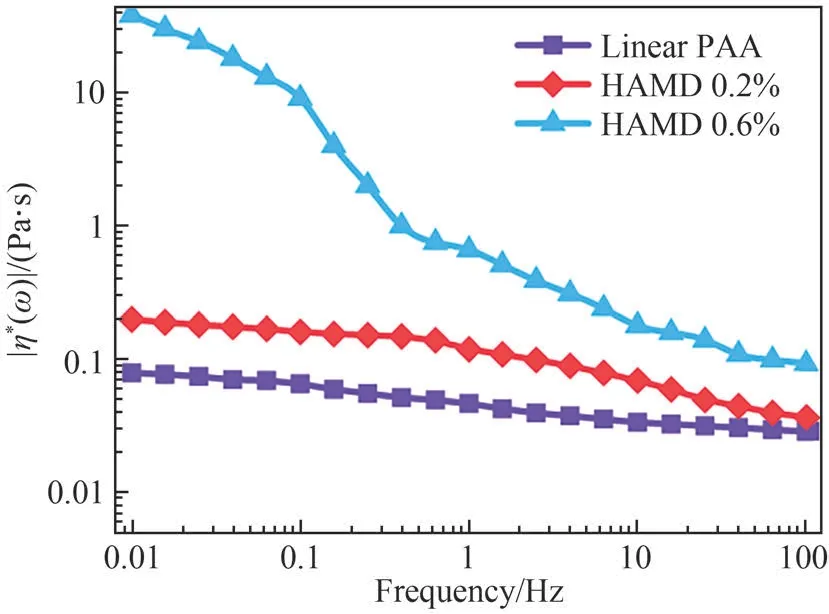
Fig.2 Shear rate-dependence of viscosity for linear PAA polymer chains modified with different hydrophobic branches,and distinctive configurations
Because liquid-like and gel-like behaviors can be distinguished from the frequency-dependence of the storage modulus (G') and loss modulus (G")[14], herein,the onset of the gel point during the gelation process was determined by applying the rule that the values ofG' andG" follow the same frequency-dependence(G' ≈G" ≈ωn) at this point.The typical gelation process is illustrated in Fig.3(a) for the system containing 10 wt% hydrophobic branches: the solution initially behaved like a low-viscosity liquid (G'

Fig.3 (a)Typical evolution of elastic modulus,G'(open symbols),and loss modulus,G"(full symbols),as a function of frequency during the gelation process(HAMD 10%);(b)phase angle δ versus time for the systems with different HAMD concentrations(1 Hz)
It is well known that the rheological behavior during the gelation process can be characterized in terms of a simple relationship (Eq.(1)) between the dynamic moduli (G' andG") and phase angle,δ(which is proportional to the value of the viscoelastic exponent,n,and independent of the angular frequency)[15-16].
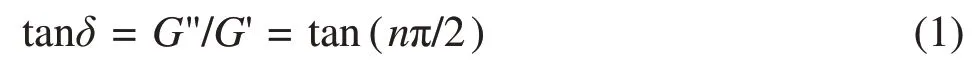
At the gel point, the following relation describes the incipient gel:

To interpret the evolution of the viscoelasticity during gelation,the complex viscosity is given by:
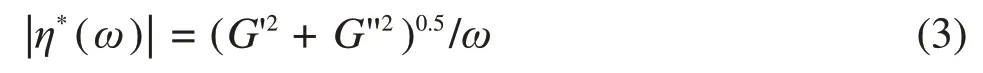
Therefore, the frequency-dependence of the absolute value of the complex viscosity can be described by the power lawη*∝ω-m(Fig.4(a)), where the complex viscosity exponentmfor an incipient gel is directly related tonbym=1-n.Herein,a value ofmclose to zero indicates liquid-like behavior, whereas a value ofmapproaching 1 indicates solid-like behavior of the system.
The frequency-dependence of the complex viscosity at different stages,Φ(Φ= (t-tg)/tg,tis the observed time,tgis the gelation time, andΦis the relative distance from the gel point) during the physical gelation process for aqueous solutions with different hydrophobic branches and CNC concentrations is shown in Fig.4(b).The general trend was parallel for all samples, that is, at the early stage of the pregel region, a negative or very low value ofΦwas found(liquid-like behavior).However, as time progressed(Φ> 0), the gelation phenomenon emerged, and a progressive increase inmwas observed, indicating solid-like behavior(mis close to 1 atΦ≫0).This result indicates that a large number of entangled and hydrophobic-associated polymer chains contributed to the elastic characteristics of the networks[17].
Far beyond the gel point (Φ> 0), |η*(ω)| became clearly higher during the gelation process (Fig.4(a)).During this stage,both inter-polymer and intra-polymer entanglement and hydrophobic association between the chains were formed,a situation in which higher density and greater rigidity of the cross-linking network would be attained[16].Fig 4(b) shows the trend of the complex viscosity exponent,m, during the gelation process for the systems with various compositions.Over the entire observed reaction course, the values ofmat a givenΦ(both in the pregel stage whereΦ <0 and in the postgel stage whereΦ >0) increased with increasing HAMD content.For the later mature stage of the gelation process (Φ> 3.5), the difference between the values ofmwas negligible for the systems with different compositions; far beyond the gel point, the HAMDs act as stickers and form intermolecular bridges that play a role similar to that of the CNCs;thus,the value ofΦis almost the same.In fact,far into the post-gel region, most of the hydrophobic associative sites on the polymer chains act as analogous cross-links and lead to the formation of strongly cross-linked networks; thus, during the post-gel stage, the complex viscosity shows less frequency-dependence.
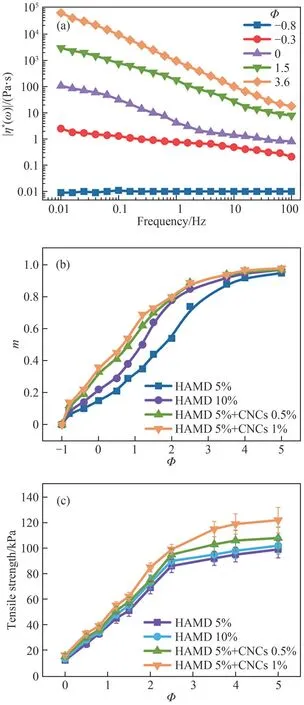
Fig.4 (a) Typical curves of frequency-dependence of complex viscosity at different stages during the gelation process(HAMD 5% + CNCs 0.5%); (b) complex viscosity exponent;and (c) tensile strength of gel at different stages during the gelation process for various HAMD and CNC compositions
Furthermore, as indicated in Fig.4(c), the incipient gel network was reinforced by the incorporation of the hydrophobic branch and CNC fraction.During gelation,the elastic response increased, and this trend became more noticeable as the hydrophobic branch and CNC content increased.For the incipient stage(Φ=0.5),the tensile strength was 22 and 31 kPa for the samples with HAMD contents of 5% and 10%, respectively.As time progressed (Φ= 2), the strength increased to 68 kPa and 76 kPa, respectively.Nevertheless, compared with the CNCs involving dual cross-linked networks, the tensile strength of the single HAMD systems was still much lower.For example, atΦ= 3.5, the tensile strength was 97 kPa and 107 kPa for HAMD 5% and HAMD 5%+CNCs 0.5%, respectively.This difference indicates that the elastic response of the HAMD incipient gel network is relatively poor and the network is fragile because of the compact conformation and numerous hydrophobic branches buried inside the coiled structures; thus, the contribution of intermolecular hydrophobic interactions to the network rigidity is poor[17].However, the HAMD + CNC dual network was strong owing to additional CNC-induced polymer chain entanglement[13].One can visualize a situation where the close-packed CNCs are interconnected by the polymer chains and the chains intertwine and entangle at a higher level to form a network; thus, the evolution of the gel network is more stable for this transient cross-linked network, even in the absence of an organic cross-linking agent[18].The difference in the tensile strength further verifies that a more "rigid" gel network is formed, consisting of both weak (hydrophobic interactions of the connected HAMDs) and strong (CNCs) domains among the neighboring chains.
3.2 Model of gelation
It is generally accepted that the formation of a physical gel network through the association of monomers or macromolecular precursors is facilitated by noncovalent interactions such as hydrogen bonding, metal coordination,or hydrophobic interactions[8].The ability to form such non-covalent associations depends on many parameters, such as the polymer concentration,temperature, pH value, molecular weight, and type of solvent used[8,14].As an example, the current hydrophobically modified system can form associative domains with functionality higher than two and undergo gelation transition through the evolution of apercolate structure at a critical concentration[13].In contrast to their corresponding chemically cross-linked counterparts, physical polymer networks generally exhibit complex nanostructures at different levels through the inter-chain bundling,stacking,or clustering of transient cross-links[8].
Overall, the above rheological analysis indicates that the formation of cross-linked polymer networks occurs under favorable conditions, such as in the presence of sufficiently high concentrations of the monomer or analogous cross-linking agent.The gelation process is usually signaled by a strong increase in the sample viscosity, and the formation of a network is facilitated thereafter.In contrast, low viscosity and fluid-like behavior are commonly observed when network formation is suppressed.A model of the physical gelation process based on the rheological measurements is shown in Fig.1.In the early stage (Φ< 0), the system is a free-flowing liquid with the appearance of a solution, comprising small, separated, CNC-rich particles/hydrophobic association clusters.These particles then become increasingly interconnected until the gel point (Φ= 0), where the amount of microgel particles is sufficient for them to aggregate and randomly form a percolating network (Fig.5).There was no bulk hydrogel formation during the initial gelation process (Fig.5(a)), where only some freemoving CNC-PAA precursors were present in the network.As the gelation process continued, the initial free-moving clusters formed entangled networks(Fig.5(b)), which contributed to the mechanical reinforcement of the hydrogels.In fact,the dilute phase and gel phase co-exist in a two-phase equilibrium: if the attraction between polymer chains, entanglement,or hydrophobic association is strong(the corresponding characteristic energy per chain due to the formation of some physical entanglements or hydrophobic association domains,ε, is much higher than the system thermodynamics of kinetic energy,ε>ΚT),the polymer chains aggregate spontaneously and form a gel network, where the system is similar to a chemically cross-linked system.In the other case,if the associative energy is weak (ε<ΚT), the free-moving polymer chain clusters spread throughout the entire solution and no gel can be formed[19].

Fig.5 TEM images of hydrogels (HAMD 5% + CNCs 1%) at different gelation stages(a)Φ=-0.3 and(b)Φ=1.5,where the initially isolated particles form a close,entangled structure
3.3 Heat-induced dissociation and reversibility
To study the thermally induced gel-sol transitions, a swollen gel with a water content of 2000 wt% was prepared and gradually heated to examine the phase transitions.At 25℃, the sample was a clear, freestanding gel, which remained immobile under its own weight when inverted.Upon increasing the temperature to 55℃,the sample began to flow under its own weight when tilted, but largely retained a bulk shape.With a further increase in temperature, the gel turned into a clear, free-flowing liquid with low viscosity.The dissolution curves of the associative gel in Fig.6 were determined by the combination of the glass-tube inverting method and mass loss ratio test (temperature range of 55℃-75℃) for two samples with different hydrophobic branch content; a large part of the closepacked polymer domains broke from the associative gel and moved to the dilute solution,indicating that the associative gel dissolved as the cross-linked network disappeared, finally resulting in bulk dissolution of the associative gel.Moreover, it was noted that the volume of the physical gel underwent a sharper contraction with a decrease in the content of the hydrophobic fraction; because the attraction force becomes weak,some grafted chains move more freely in the dilute solution rather than remaining in the original concentrated gel network; thus, the polymer fraction in the solution phase increases.
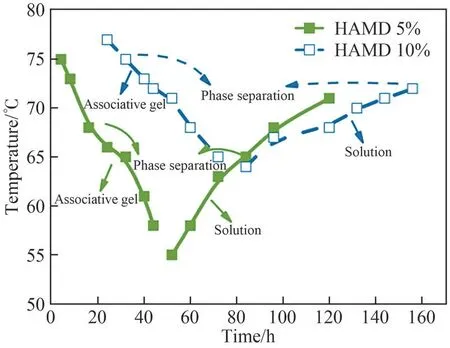
Fig.6 Dissolution curves for associative hydrogels with different contents of hydrophobic associations
Upon heating the gels towards and beyond thecritical temperature,the cross-linked structures undergo rearrangement and eventually decompose.For example,when the solution temperature was held at 75℃ for several hours, the entanglements and hydrophobic associations within the network disappeared (HAMD 10%), and the system reverted to the original singlephase solution with a uniform transparent appearance.The physical gels are connected by non-covalent bonds,and the network may collapse at some timescale because of external stimuli.Specifically, on a timescale shorter than the lifetime of the cross-links, the system behaves like a permanently cross-linked rubber; for longer timescales, the breakage of a few cross-links allows the chains to diffuse along the confinement of the structure; on a timescale longer than the terminal polymer chain relaxation time, the onset of chains viscous flow is observed[20].
The reversibility of the gel heat-cool cycle-induced network association-dissociation was evaluated by heating the swollen hydrogels from 25℃ to 75℃ and then cooling to 25℃.Typically, the storage modulus(G') of physical gels varies as a function of temperature,as shown in Fig.7.At low temperatures (~25℃), water molecules are presumed to form a cage-like structure that surrounds the hydrophobic domains.The elementary event in the dynamics of associating polymers is the dissociation of clusters from the original aggregate upon heating and re-association with reversible aggregation upon cooling.Upon heating,these regular structures distort and break, exposing the hydrophobic regions, which triggers network dissolution.Upon cooling, the damage to the network is almost an irreversible process for the hydrogel network formed by the single CNC-induced entanglement, where the solution state persists when the system is cooled to a low temperature (25℃).However, for the gels with HAMD, the network could partially revert to its initial cross-linked state, where the value ofG' after cooling was approximately 70% of that of the native gel.This reversible association normally occurs in semi-dilute systems or systems with high concentrations and disappears via dissociation as the solution is diluted or heated/cooled again; that is, the reversible dissociationassociation of the cross-link junctions in the gel network contributes to the recovery behavior.However,irreversible aggregation (CNC-induced entanglement)leads to a metastable colloidal structure during the dissolution process.This result suggests that when HAMDs are introduced into network structures, the formation of cross-links is reversible because of the local solubilization of the hydrophobic interactions, and the network is more viscoelastic owing to the weak interactions.Because the thermally triggered dissolution of the micelle around the hydrophobic blocks may facilitate the hydrophobic interactions within the local range, whereas the conformational rearrangement woulddecrease these interactions again, it is likely that the micellar kinetics and resulting temporary strong associations are responsible for the network reversibility[21].

Fig.7 Typical thermoreversibility of storage modulus(G')as a function of temperature at a frequency of 1 Hz.The line with arrow represents the heating and cooling curve.After the heatcool cycle, the HAMD+CNC induced gel survives the heattriggered dissociation and partially recovers its original aggregated state, whereas the original entanglement in the single CNC-induced gel is broken irreversibly.
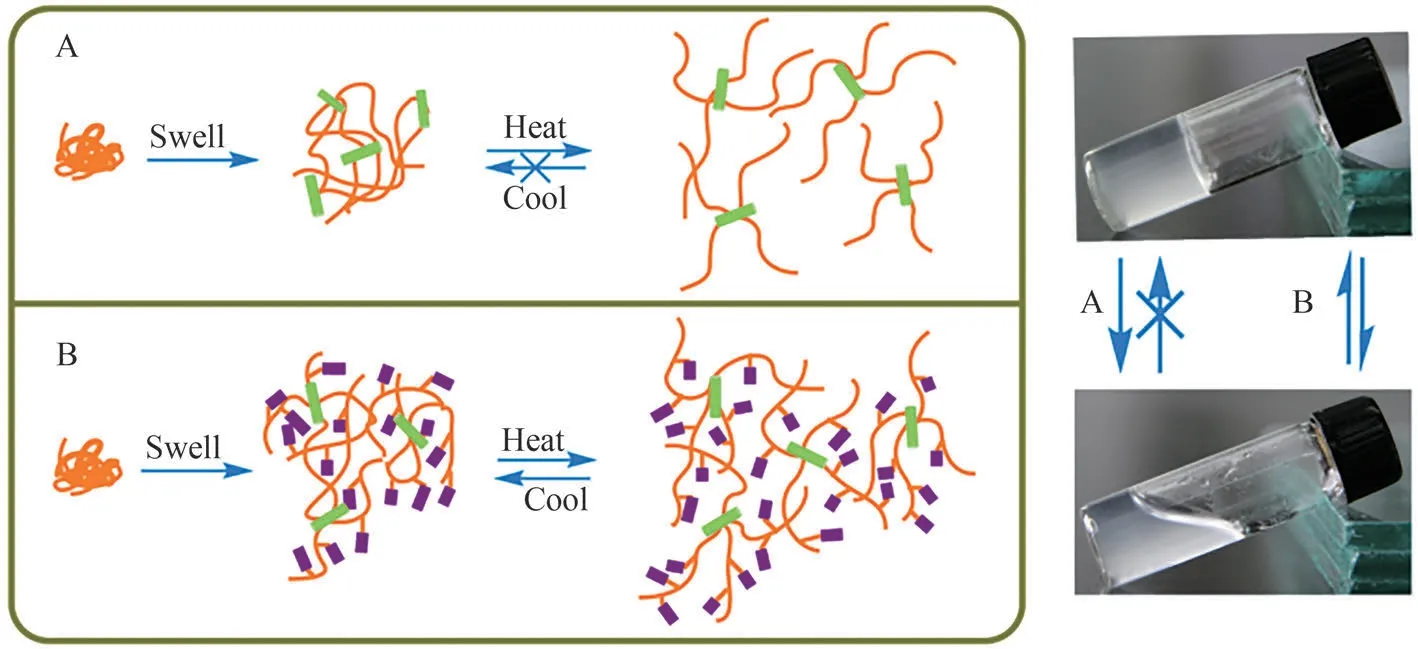
Fig.8 Schematic of elementary steps for gel-sol transition in two types of polymer networks (CNCs (A) and HAMD+CNCs(B)).By raising the system temperature, the thermal energy breaks the association and entanglements, thereby forming the sol state.The process is reversible for the latter system (B), which partially recovers from sol to gel because the hydrophobic associations dominate the re-association process at low temperatures.However, in the single CNC-induced system (A), transition of the sol state to the gel state is irreversible once the polymer chains undergo disentanglement.
4 Conclusions
The formation and collapse of cellulose nanocrystals and hydrophobic association-induced hydrophilic/hydrophobic systems were examined through rheological measurements.At low concentrations, the hydrophobic association domains form clusters that contain several backbone chains, and an increase in the concentration of hydrophobic association micro-domains (HAMD)increases the number and size of such clusters, which significantly facilitates the formation of interconnected networks.This indicates that the addition of cellulose nanocrystals to the dilute polymer solution favored the formation of associative gels.During the gelation process, a strong enough attraction between the hydrophobic domains promotes the establishment of a gel-like network between a fraction of the macromolecular precursors;in the other case,only freemoving chains or clusters are formed, in which the fraction of the associative gel phase is far from unity and can be neglected.Analysis of reversible associationdissociation in the network demonstrated that the fraction of hydrophobic branches in the polymer contributes to the temperature sensitivity of the gel and gel-sol-gel transition, where this thermally induced rearrangement of the polymer chains leads to hydrogels having potential applications in stimuli responsive materials.
Acknowledgments
This study was financially supported by the College Student Research and Career-creation Program of Beijing(S202010022195).
杂志排行

Paper and Biomaterials的其它文章
- Nanocomposite Hydrogel Materials for Defective Cartilage Repair and Its Mechanical Tribological Behavior—A Review
- Review of Carbon Dots from Lignin:Preparing,Tuning,and Applying
- Octadecylamine Graft-modified Cellulose Nanofiber and Its Reinforcement to Poly(butylene adipate-co-terephthalate)Composites
- NIR-responsive Collagen-based Sponge Coated with Polydimethylsiloxane/Candle Soot for Oil-Water Separation
- Stabilizing Pickering Emulsions Using Octenyl Succinic Anhydride Modified with Cellulose Nanofibrils
- Technical Evaluation of Hybrid Clones of Corymbia spp.to Produce Market Pulp