电子束选区熔化成形Ti2AlNb合金微观组织与性能
2022-07-18车倩颖贺卫卫李会霞程康康
车倩颖,贺卫卫,2*,李会霞,程康康,王 宇
(1 西安赛隆金属材料有限责任公司,西安 710018;2 西北有色金属研究院 金属多孔材料国家重点实验室,西安 710016)
Ti2AlNb基合金是一种以O相有序正交结构为基础的金属间化合物材料,因具有优异的室/高温抗拉强度、疲劳强度,良好的断裂韧度和抗蠕变性,以及低的热膨胀系数等优点,近年来成为研究的热点。Ti2AlNb基合金服役范围在600~800 ℃,是替代镍基高温合金最具潜力的材料[1-2]。然而Ti2AlNb基金属间化合物加工能力差,铸造、锻造、粉末冶金传统制备方法存在材料利用率低、制造工艺复杂、开发周期长等问题,很大程度上限制了Ti2AlNb合金的实际应用[3-5]。增材制造(additive manufacturing,AM)是基于CAD模型的分层制造技术,可以制备出具有任意复杂形状的零件,无须模具,工序简单,很大程度上缩短了生产周期,降低了成形件的设计和制造成本,被认为是最有发展前景的制造工艺[6-7]。近年来,关于γ-TiAl基合金的AM研究较多[8-10],而针对Ti2AlNb基合金AM的报道较少。Polozov等[11-12]采用Ti粉、Al粉、Nb粉以激光选区熔化成形(selective laser melting,SLM)技术原位合成Ti-22Al-25Nb合金,然而合金内存在大量微裂纹、气孔和部分未熔化的Nb元素,导致其力学性能较差,而要完全熔化Nb元素需要更高的能量,但这会导致Al元素的损失,因此Nb元素和Al元素之间的协调关系还需进一步研究。唐杨杰等[13]利用基于同轴送粉方式的激光熔化沉积技术(laser melting deposition,LMD)制备了Ti-22Al-25Nb试样,该沉积态试样显微组织沿高度方向呈现出O+α2+B2→O+B2→B2的变化趋势,沉积态试样室温和750 ℃抗拉强度最高为1053 MPa和665 MPa,断后伸长率分别为2.8%和1.3%。Zhou等[14-15]采用SLM制备了Ti-22Al-25Nb合金,分析了沉积态的微观组织演变、相变规律和力学性能强化机理,并研究了工艺参数对其影响规律,发现搭接间距增加导致晶粒细化、位错密度增加和O相含量增加,提高室温抗拉强度,但过大的搭接间距会使层间结合力差,显著降低力学性能。
目前Ti2AlNb基合金的增材制造主要采用激光增材制造技术,由于SLM技术无法使粉床升温至较高温度,快速熔凝Ti2AlNb合金在成形后存在较大热应力,组织内部易产生微裂纹,在成形或后续使用过程中扩展为宏观裂纹,最终导致零件过早失效[12],而LMD较SLM熔池大,冷却速率低,但低功率时试样内部仍出现裂纹,且表面质量较差[16]。相对于激光增材制造技术,电子束选区熔化(selective electron beam melting,SEBM)技术具有更快的扫描速度,成形效率高;同时粉末熔化成形温场较高,可有效减缓成形件的热应力;另外,SEBM技术真空环境有效控制对氧和氮等杂质的吸收[17-18]。因此,SEBM在开发脆性材料方面具有较大的优势。目前采用SEBM成形Ti2AlNb基合金方面的研究鲜有报道,本工作开展SEBM成形Ti2AlNb基合金的工艺开发,系统分析SEBM成形Ti2AlNb基合金显微组织、物相演变和力学性能,以及热等静压(hot isostatic pressing,HIP)对其组织、物相和性能的影响。
1 实验材料与方法
SEBM用Ti2AlNb合金粉末采用气雾化方法制备,合金的名义成分为Ti-22Al-25Nb,实际化学成分如表1所示。图1为Ti-22Al-25Nb合金粉末形貌扫描照片,可以看出,粉末呈球形,有少量卫星粉,其粒度范围为53~150 μm。

表1 Ti-22Al-25Nb粉末和成形试棒化学成分Table 1 Chemical compositions of the Ti-22Al-25Nb powder and as-built rods

图1 Ti-22Al-25Nb合金粉末形貌Fig.1 Morphology of Ti-22Al-25Nb powder
Ti2AlNb合金的SEBM工艺开发采用西安赛隆金属材料有限责任公司生产的S2型SEBM装备,实验用成形底板为316L不锈钢,尺寸为120 mm×120 mm×10 mm,在真空度不大于5×10-2Pa的真空成形室中,利用电子束将基板预热到850~900 ℃,然后在成形底板上铺展0.05 mm层厚的粉末,再以电子束预热粉末,粉末预热完成后,根据零件截面信息进行选区熔化,选区熔化电流为11~14.5 mA,熔化速度为2800~4700 mm/s,熔化完成后成形底板下降一定高度,重复上述过程,完成整个试样打印,如图2(a)所示。随后对制备的部分试样进行热等静压处理,热处理制度为压力150 MPa,温度1030 ℃,保温保压3 h。
成形柱状试样沿沉积方向间隔5 mm切取试样块(图2(b)),从上到下分别命名为试样1,试样2,试样3,试样4,试样5,对应高度分别为25,20,15,10,5 mm。采用AXIO Vert A1型光学显微镜(OM)和JSM-6460型扫描电子显微镜(SEM)观察分析试样微观组织,显微组织试样采用砂纸打磨至2000#后进行抛光,然后以1 mL HF+3 mL HNO3+7 mL H2O腐蚀液进行腐蚀,时间为15~20 s。采用XS205型电子天平测试试样的密度。采用Bruker D8 Advance型X射线衍射仪(XRD)进行物相分析,Cu靶,衍射范围为20°~90°,步长0.02°,每步0.5 s;采用MVS-1000IMT2型显微硬度仪测试试样的显微硬度,载荷为1.96 N,加载时间10 s。切取横向拉伸试样(图2(c)),在INSTRON 5982型拉伸试验机上进行室温拉伸实验,试样标距为25 mm,拉伸速率为0.1 mm/min,至少测试3个样品。采用SEM观察拉伸试样的断口形貌。

图2 SEBM成形Ti2AlNb试样照片(a),不同高度显微组织取样示意图(b)及拉伸试样图(c)Fig.2 Photograph of the Ti2AlNb samples by SEBM (a),schematic diagram of microscopic tissue sampling at different heights (b) and tensile samples (c)
2 结果与分析
2.1 致密度优化
增材制造工艺中,适宜的热源能量密度输入是获得表面平整光滑、组织致密零件的关键。SEBM技术常采用电子束线能量密度评价工艺参数对成形试样致密度的影响,线能量密度(LE)计算公式表示为:LE=I×U/(v×h×t),其中,I为熔化电流,mA;U为电子束高压,60 kV;v为熔化速度,mm/s;h为熔化间距,mm;t为粉层层厚,mm。本研究固定h为0.1 mm,t为0.05 mm。图3为SEBM成形Ti2AlNb合金的工艺窗口,从图3可以看出,在过高及较低的线能量密度输入下都无法获得表面质量良好的试样。线能量密度为48~52 J/mm时,能量输入过高,导致熔池尺寸增大,液相较多,易使成形件表面凸起,而不平滑的表面会使铺送粉末层厚不均匀,导致粉末熔覆过程层间结合不均匀,影响致密度[19];线能量密度小于36 J/mm时,较快的扫描速度或较低的熔化电流使得热输入不足,导致粉末不能完全熔化,熔池无法完全铺展,层间结合差,内部容易形成孔隙或裂纹,表面留下较多的孔洞,成形质量较差;线能量密度为36~48 J/mm时,电子束热量输入与粉末熔化速度匹配良好,打印工艺执行顺利,最终成形试样表面平整。
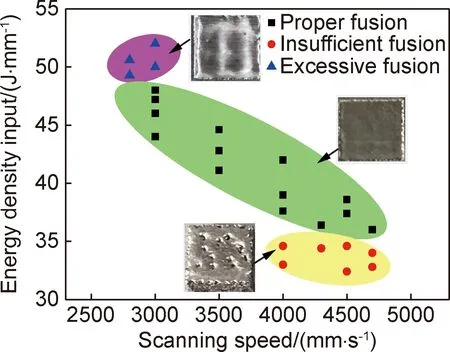
图3 SEBM成形Ti2AlNb合金的加工窗口Fig.3 Processing window for fabricating Ti2AlNb alloy by SEBM
致密度会直接影响成形试样的力学性能。在成形表面质量良好的工艺窗口内,选择线能量密度在36~48 J/mm之间的一组工艺参数进行打印,测试打印试样块密度,并分析其内部缺陷。图4为不同线能量密度打印试样块与密度的关系图,当线能量密度为36~38 J/mm时,密度测试结果在5.37~5.39 g/cm3之间,这类试样内部缺陷为细长的未熔合孔洞,这是由于低能量密度下层间结合不足形成的缺陷。随着能量密度的增加,密度值也逐渐增加,当线能量密度为40~43 J/mm时,可获得内部致密基本无缺陷的试样,密度测试结果为5.42~5.43 g/cm3。进一步提升线能量密度,熔化区域积累的能量增加,导致上层已凝固的粉末发生重熔,另外较大电子束流作用易引起粉末飞溅,致使熔池表面生成夹杂缺陷,降低试样的致密度。

图4 线能量密度与试样密度的关系Fig.4 Effect of line energy density on the relative density of the samples
2.2 化学成分
采用熔化电流为12.5 mA,熔化速度为3500 mm/s(线能量密度为42.9 J/mm)的工艺制备Ti2AlNb合金试样,该试样的化学成分如表1所示,可以看出,与粉末相比,成形试样中O,N,H等间隙元素含量未有明显变化,但Al元素出现1.2%的烧损,Nb元素略有增加。SEBM成形过程是在高真空环境中进行,可以减少O,N,H等杂质元素的污染。但由于电子束能量密度高,熔池中心温度较高,导致蒸气压高的Al元素因蒸发而损失[20]。根据SEBM成形Ti-48Al-2Cr-2Nb合金的研究,可通过减小能量输入、降低扫描间距或合理规划扫描路径减少Al损失[16]。
2.3 显微组织演变
沉积态Ti2AlNb合金试样内部致密,只存在极少量微小孔隙,试样密度值为(5.42±0.006) g/cm3。HIP后试样内部孔隙闭合,但密度值((5.42±0.005) g/cm3)较沉积态变化不大,一方面是由于沉积态试样缺陷极少,另一方面主要是由于排水法测试密度对少量微小孔洞具有较低的敏感度,所以热等静压前后试样的密度差别不大。图5是Ti2AlNb合金沉积态(左侧)和热等静压态(右侧)试样在不同高度纵截面显微组织金相照片。从图中可以看出,沉积态纵截面显微组织呈现柱状晶结构,宽度方向的平均晶粒尺寸为(104.02±32.78) μm,SEBM成形过程中沿沉积方向温度梯度较大,使得晶粒沿着沉积方向外延生长形成柱状晶结构。这种外延生长导致的柱状晶结构在激光选区熔化[21]和激光熔化沉积[22]等增材制造方法中也有发现。图5(a-1)为沉积态试样顶部组织,板条析出相的数量较少,而越靠近底板,析出相的数量越多(图(b-1)~(e-1)),主要是由于已成形的材料受到上方熔池的影响而产生周期性的加热、冷却和凝固,随着成形高度的增加,底部材料经历的热循环次数逐渐增多,导致析出相增加[13]。与本研究结果不同的是,Tang等[23]的研究显示沉积态试样的底部主要由O+α2+B2三相组成,顶部则主要由B2相组成,这是顶部成形结束后较高的冷却速率抑制了B2相向α2相和O相转变引起的。同样SEBM成形也具有快速加热和冷却的特点,但在顶部却出现少量的析出相,这主要是由于SEBM成形的温度场较高,驱动B2相发生转变,导致α2相和O相析出。另外,成形试样中可以观察到层层堆叠的由于光斑能量高斯分布引起的弧形熔池轮廓,如图5(e-1)中箭头所示,且熔池内部的晶粒尺寸极细。HIP后试样组织结构呈现近柱状晶特征,沿沉积方向的晶粒尺寸明显减小,宽度和长度方向的平均晶粒尺寸分别为(105.84±32.67) μm和(282.26±91.03) μm。另外从顶部至底部的显微组织呈均匀化分布,这主要与HIP过程中高温和高压作用有关,且析出相的数量相当,但相比沉积态而言,析出相的宽度缩小且数量明显减少(图5(a-2)~(e-2))。

图5 沉积态(1)和热等静压态(2)Ti2AlNb合金不同高度处显微组织(a)试样1;(b)试样2;(c)试样3;(d)试样4;(e)试样5Fig.5 Microstructures of as-built (1) and HIP (2) Ti2AlNb alloy at different heights(a)sample 1;(b)sample 2;(c)sample 3;(d)sample 4;(e)sample 5
试样1~5显微组织类似,本工作选用试样3进行SEM分析。图6为沉积态和HIP态试样的SEM组织,均由B2基体、晶内尺寸较大的板条状初生O/α2相和细小的次生针状O相组成,析出的次生O相排列杂乱无章,尺寸约1 μm,初生板条相呈魏氏体分布,沉积态板条相的平均长度为6~12 μm,宽度在4 μm左右。HIP后,O/α2相的宽度和数量相对沉积态明显减少,由Ti2AlNb伪二元相图可知[24],1030 ℃的HIP温度处于α2+B2双相区,在此温度下进行加热和保温,晶粒内部分O相和α2相溶解于B2基体,相变过程为O→α2+B2,O+α2+B2→α2+B2。而在随后的冷却过程中O相充分形核,发生B2→O+B2相变,随着缓冷过程中温度的降低,O相长大的动力学条件受限,形成细小的针状O相分布于B2晶粒内[25]。

图6 沉积态(a)和HIP态(b)Ti2AlNb合金显微组织SEM照片Fig.6 SEM images of as-built (a) and HIP (b) Ti2AlNb alloy
2.4 物相分析
图7为沉积态和HIP态不同高度Ti2AlNb合金试样的X射线衍射谱,由图可知,Ti2AlNb合金试样沉积态和HIP态均由B2,O和α2相组成。沉积态和HIP态试样不同高度处B2和O相的衍射峰强度较高,α2相衍射峰强度较低,表明含有少量的α2相。随着打印高度的增加,沉积态试样下部材料的O相衍射峰增强,这是由于已沉积材料受上部熔池的影响发生周期性加热、冷却,使已沉积层进行自时效处理,B2相转变成O相,使得O相衍射峰强度增加,与显微组织分析结果相符。相比沉积态试样4和试样5,HIP态试样由于板条析出相体积分数的减少导致O相衍射峰强度的降低,同时不同高度处的物相分布及衍射峰强度基本一致,与HIP后自上而下组织均匀化的结果相符。

图7 沉积态(a)和HIP态(b)试样不同高度处的XRD图谱Fig.7 XRD patterns of as-built (a) and HIP (b) samples at different heights
2.5 硬度分析
图8为沉积态和HIP态试样不同高度处的显微硬度值。结果显示,沉积态的硬度值随着成形高度的增加从(345.87±5.09)HV降至(294.33±5.29)HV,硬度值的变化与显微组织有明显的对应关系,沉积态试样顶部组织以基体B2相为主,而B2相为体心立方塑性相,抵抗变形能力较差,导致顶部硬度值较低。随着试样高度逐渐增加,下部已沉积材料经历周期性的加热、冷却,在多次的热循环作用下,α2/O板条析出相的数量逐渐增加,析出强化作用使得硬度值升高[26]。HIP后显微硬度值增加至388.91~390.48HV之间,且不同高度处的硬度值相差不大,这主要与HIP后显微组织析出相的均匀化有关(图5),且大量细小针状O相的析出和α2/O相宽度的减小,提高了合金的显微硬度。
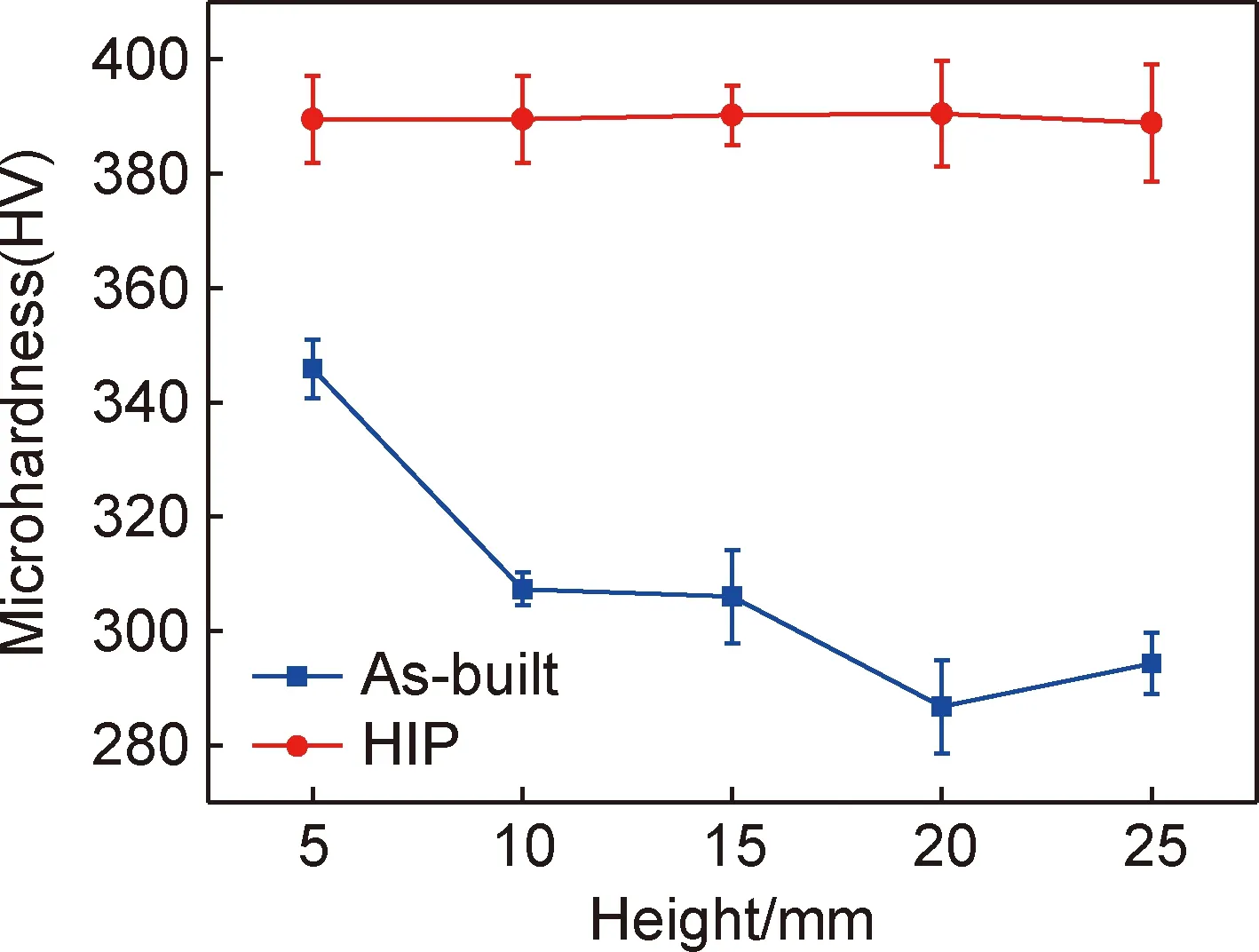
图8 沉积态和HIP态试样不同高度处显微硬度Fig.8 Microhardness of as-built and HIP samples at different heights
2.6 力学性能
图9为Ti2AlNb合金沉积态和HIP态的室温力学性能,可以看出沉积态试样室温抗拉强度为(1061±23.71) MPa,屈服强度为(890±42.77) MPa,伸长率为(3.67±1.15)%,HIP后抗拉强度和屈服强度分别增加至(1101±23.07) MPa和(934±43.39) MPa,但伸长率稍有下降,降至3.5%。合金的拉伸性能主要受晶粒尺寸和析出相的影响,HIP后柱状晶粒平均尺寸减小,根据Hall-Patch关系,细小的晶粒尺寸可以提高合金强度;另外,初生板条析出相的宽度明显缩小,且数量减少,晶内二次针状O相的增加使O相与B2相基体的相界面增加,提高了合金强度,但也降低了塑性[18]。唐杨杰等[13]采用LMD成形Ti-22Al-25Nb合金的最高室温抗拉强度和伸长率分别为1053 MPa和1.3%,相比之下本研究成形试样的强度和塑性均得到改善。与Zhou等[15]的研究结果进行对比,发现成形试样的抗拉强度相差不大,但Zhou等的伸长率更高,主要是与SLM超快冷却速度抑制脆性α2相的形成有关。α2相为密排六方结构,滑移系最少,变形较为困难,不利于伸长率的提高,因此抑制α2相形成有利于合金强度和塑性的提高。另外,对比Ti2AlNb合金的显微硬度值,发现硬度较强度的提升效果显著,这可能与电子束成形后试样内部形成的低位错密度有关[25],导致合金的强度有所降低,而细化的晶粒尺寸和析出相宽度均有利于强度增加,因此,在两者的综合作用下,HIP后合金的强度提高较少。基于目前SLM和SEBM成形Ti-22Al-25Nb合金的研究较少,针对获得优异综合性能的成形试样还需开展进一步研究。

图9 Ti2AlNb合金室温拉伸性能Fig.9 Tensile properties of the Ti2AlNb alloys at room temperature
图10为Ti2AlNb合金沉积态和HIP态拉伸断口形貌,从图10(a)可以看到解理台阶和河流状花样,为典型的解理断裂,同时断口上观察到了少量的韧窝,意味着断裂前合金发生了塑性变形,因此沉积态试样呈现出解理断裂和韧性断裂在内的准解理断裂特征。HIP后合金的断口形貌与沉积态相近,此外,在沉积态和HIP态试样断口中都存在明显的板条状组织被剥落留下的痕迹,而板条尖端的应力随着板条尺寸的减小而提高,应力集中易在板条尖端处产生,HIP后板条组织的宽度减小,导致试样过早开裂失效,降低了伸长率。

图10 拉伸断口SEM照片 (a)沉积态;(b)HIP态Fig.10 SEM morphologies of fracture surface (a)as-built;(b)HIP
3 结论
(1)电子束选区熔化成形Ti2AlNb合金的线能量密度为36~48 J/mm时试样表面质量良好,线能量密度优选40~43 J/mm范围内试样密度可达到5.42~5.43 g/cm3。
(2)Ti2AlNb合金沉积态和HIP态的组织均为柱状晶结构,沉积态试样宽度方向的平均晶粒尺寸为(104.02±32.78) μm,HIP后长度方向的晶粒尺寸减小,宽度和长度方向的平均晶粒尺寸分别为(105.84±32.67) μm和(282.26±91.03) μm。
(3)沉积态和HIP态试样的显微组织均由B2基体、晶内尺寸较大的板条状初生O/α2相和细小的次生针状O相组成,从顶部到底部沉积态试样的板条状初生O/α2相逐渐增多,HIP后组织趋于均匀化,且初生O/α2相的宽度缩小,数量减少。
(4)沉积态试样的显微硬度值随着成形高度的增加而减小,最高值为(345.87±5.09)HV,HIP后显微硬度值增加至388.91~390.48HV之间。
(5)沉积态合金抗拉强度为(1061±23.71) MPa,伸长率为(3.67±1.15)%,HIP后合金抗拉强度增加至(1101±23.07) MPa,伸长率稍降至3.5%。细晶强化和相界面增加是提高合金强度的主要原因。
