基于GEO数据库筛选肺炎链球菌感染的关键基因和信号通路研究
2022-07-17李静,王书,张颖
李 静,王 书,张 颖
(安徽医科大学第三附属医院(合肥市第一人民医院) 老年医学科,安徽 合肥 230000)
0 引言
肺炎链球菌是一种人类共生细菌和主要的机会性呼吸道病原体,它不仅会导致严重的侵袭性疾病,甚至每年在全世界造成数百万人死亡[1]。鼻咽分泌物的吸入可使肺炎链球菌在肺实质中侵袭和传播,从而导致肺部感染[2]。肺炎球菌肺炎的高发病率和高死亡率给个人和社会带来了沉重的医疗负担[3]。因此,探究机体对肺炎链球菌感染的反应机制对防治肺炎链球菌感染至关重要。近年来,运用生物信息学方法在分子水平上进行数据挖掘,为研究各种疾病的分子机制提供了新的思路。本研究通过对肺炎链球菌感染小鼠的肺关键基因的分析及鉴定,以期了解机体抵制肺炎链球菌感染背后的分子机制。
1 资料与方法
1.1 数据下载和处理
GSE83612数据集来自GEO数据库(https://www.ncbi.nlm.nih.gov/geo),共纳入4例肺炎链球菌(SP)感染小鼠和4例磷酸盐缓冲盐水 (PBS)对照的小鼠基因表达信息,其检测平台为GPL6887。对从 GEO 数据库下载的探针表达矩阵文件进行归一化和log2转换,将平台注释文件与每个探针表达矩阵进行匹配,并保留注释良好的探针。对于对应一个基因的多个探针,取平均表达值用于进一步分析。
1.2 DEG 的鉴定
基于对照(HC)样本和SP样本表达值的比较,通过RStudio软件(版本:1.3.1056)的limma包[4]筛选出差异表达基因(DEGs)。DEG 的筛选标准如下:log2 倍数变化 (FC) 应大于 2 或小于-2,调整后P<0.05。
1.3 功能富集分析
应用colorspace,stringi,colorspace包对差异表达基因进行 GO (Gene Ontology)分析和 KEGG (Kyoto Encyclopedia of Genes and Genomes)通路分析,P<0.05且Q<0.05为筛选条件。
1.4 PPI网络构建与模块分析
为了探索不同基因编码蛋白质之间的相互关系,将DEGs导入STRING网站(https://www.string-db.org/)进行进一步分析。最低交互分数应大于 0.9,并删除网络中的孤立节点。接下来,将分析结果输出一个TSV格式的文件中,并使用Cytoscape软件(版本:3.8.2)进行细节处理和模块分析。MCODE是从 Cytoscape App Store下载的插件,可以根据拓扑结构在复杂网络中找到紧密相连的节点。因此,应用此插件,以degree cutoff=2,node score cutoff=0.2,k-core=2和max.depth=100为标准[5],来检测PPI网络中的关键模块,并应用Cytohubba插件,按degree算法计算出评分位于前10位的枢纽基因。
2 结果
2.1 DEG 的鉴定
通过使用limma包分析整合数据集的差异表达,获得了325个DEG由281个上调基因和44个下调基因组成(表1列出部分),以及按调整后的P值排序的前 20 个基因的表达(见表2)。
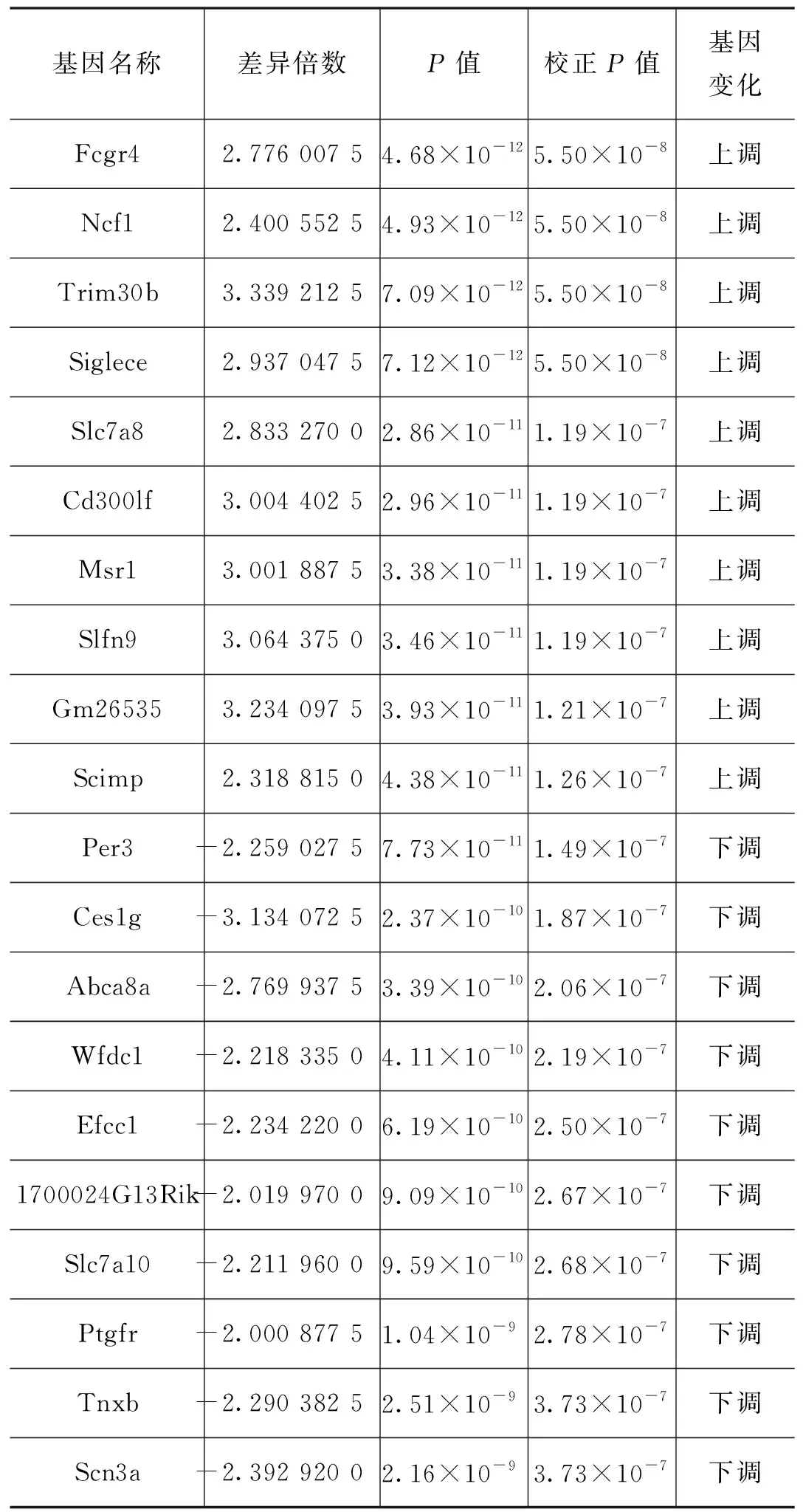
表1 部分差异表达基因

表2 按调整后的P值排序的前20个基因的表达
2.2 GO 和 KEGG 富集分析
Cluster Profiler包进行的富集分析揭示了与DEG相关的生物学功能和途径。如表3所示,DEG的GO注释由3部分(BP,CC,MF)组成(每部分列出前10项)。发现主要细胞成分(CC)位于血浆脂蛋白粒子、液泡、溶酶体等;主要分子功能(MF)与细胞因子活性、趋化因子活性、G蛋白偶联受体等相关;主要涉及白细胞及各种粒细胞迁移、γ-干扰素反应等生物过程。KEGG分析的前10条路径(见表4)表明DEGs参与信号转导和炎症反应相关通路,如肿瘤坏死因子信号通路、Toll样受体信号通路、细胞因子-细胞因子受体相互作用和IL-17信号通路等。

表3 差异表达基因的GO富集分析结果(列出了每个类别的前10项)

表3(续)

表4 差异表达基因的KEGG富集分析结果(列出前10项)
2.3 PPI2.3网络搭建与MCODE插件分析
STRING数据库构建的PPI网络经Cytoscape调整可视化。为了找出复杂网络的核心模块,进行了MCODE插件分析,确定了得分最高的2个重要模块(其一:Ccl4,Il1b,Il1a,Csf2,Ccl7,Tnf,Cxcl13,Cxcr3,Cxcl1,Il6,Cxcl10。其二:Ifi44,Oas2,Rsad2,Isg15,MX1,Oasl1,Oasl2,Rtp4,Usp18),得分均为5分,显示主要与趋化因子及干扰素反应相关。运用cytoHubba插件,利用MCC法筛选出排名前10的枢纽基因依次为Irf7,Ifit1,Rsad2,Usp18,Oas12,Mx1,Ifi44,Cxcl10,Il1b,Isg15。
3 讨论
肺炎球菌感染可导致人类高死亡率和发病率[6]。作为鼻咽共生菌群的一部分,肺炎球菌如何从共生状态转变为致病状态所涉及的宿主反应目前尚不清楚。确定宿主感染肺炎链球菌后机体的反应机制,有利于开发宿主导向的疗法和筛选测试,进行个体化的治疗。本研究旨在通过生物信息学分析来识别肺炎球菌感染的生物标志物并揭示其生物学功能,为制定相关的防治靶点提供思路。
在分析中,筛选出281个上调和44个下调的 DEG,肺炎链球菌感染和对照样本之间的变化至少为 4 倍。按调整后的P值排序的前 50 个基因的表达显示Igsf6,Ccd33,Il1b等基因在肺炎链球菌感染的肺转录组中显著高表达,表明这些基因可能参与肺炎球菌感染的发生发展。接下来,通过功能富集分析对DEGs进行注释,观察可知这些基因与细胞反应和炎症信号密切相关,例如各种炎性细胞迁移、趋化因子信号通路、肿瘤坏死因子信号通路和IL-17 信号通路。使用STRING网站和Cytoscape软件建立DEG的PPI网络。在MCODE插件的帮助下,筛选出最重要的子网络,主要集中在调控干扰素及趋化因子的基因上,类似地,最后应用cytoHubba筛选出的前10位枢纽基因,也主要与这两种基因有关。
在枢纽基因及显著模块中,干扰素相关基因的表达显著上调。据报道,I型干扰素(IFN-I)通过调节紧密连接蛋白的表达可降低上皮细胞通透性和抑制细菌迁移[7],防止肺炎链球菌局部肺部感染发展为侵袭性疾病[8]。研究表明,缺乏 IFN-I 受体的小鼠在肺部感染肺炎链球菌后显示出菌血症风险的增加[7]。类似的,II型干扰素(IFN-γ)在肺炎球菌肺炎期间可以控制传染源和调节免疫系统[9],增强高亲和力细胞表面分子FcγRI(CD64)和主要组织相容性复合体(MHC)的表达从而参与炎症反应[10]。然而,IFN信号过度活化或没有及时消除也会引起免疫系统紊乱,有可能会引发慢性炎症,因此需要表达免疫负调控因子准确、适时地调整胞内信号通路的激活,从而保证机体免疫平衡。本研究中Irf7,Ifit1,Rsad2,Usp18,Oas12,Mx1,Ifi44,Isg15作为枢纽基因,协同调控干扰素在肺炎链球菌感染期间的作用。最显著模块中的基因还包括趋化因子及促炎细胞因子,如Cxcl10,Ccl4,Cxcl1,Ccl2等趋化因子显著上调,趋化因子可与白细胞上的细胞表面受体结合,并将它们吸引到宿主感染部位[1],通过及时和协调地招募免疫细胞,在对抗微生物感染和启动组织修复方面发挥着至关重要的作用[11]。发挥重要作用的促炎细胞因子包括IL-6和Tnf等,IL-6可通过促进肺修复、上皮细胞存活和减少成纤维细胞积累,在气道细菌和病毒感染期间作为维持屏障完整性的关键免疫介质[12];肿瘤坏死因子(TNF)是免疫的中枢调节剂[13]。在实验模型中,阻断TNF-α和IL-6等细胞因子的表达或作用会削弱肺炎球菌肺炎期间的宿主防御能力,并导致细菌清除受损和存活率降低[14]。
综上,笔者整合了多种生物信息学工具,确定了10个中枢基因可能作为肺炎链球菌感染的潜在治疗靶点,在未来可通过尝试使用化学诱导物(如趋化因子)将免疫细胞募集到肺部或直接调节免疫细胞功能(调节相应细胞因子活性或信号通路的开放)等手段治疗肺炎链球菌肺炎。
