In,X(X=B,Ce)共掺杂TiO2光催化活性研究*
2022-07-08张鹏会李艳春潘凯涛胡怀生胡浩斌
张鹏会,李艳春,刘 东,潘凯涛,胡怀生,胡浩斌
(陇东学院 化学化工学院,甘肃 庆阳 745000)
0 引 言
世界人口的增长和工业的快速发展都加速了能源的消耗,同时产生的有毒、有害物质和排放的工业废弃物等导致环境污染与破坏、全球变暖和气候的反常变化[1]。光催化技术是解决这些问题的有效途径之一,其利用丰富、清洁、安全的太阳能来降解污染物,即光化学与催化剂二者结合引发光催化氧化反应[2]。1972年Fujishima等[3]发现在TiO2电极上光催化分解水的现象,标志着多相光催化研究新纪元的开启。TiO2具有良好的生物和化学惰性、很高的介电常数、高的催化活性、低成本,对光腐蚀和化学腐蚀有很好的稳定性,被认为是当前最具有应用潜力的光催化剂之一。此外,利用TiO2光催化降解有机物污染物速度快,对被降解物几乎无选择性且最终将其完全矿化,这样可减少二次污染。然而,TiO2带隙(3.2 eV)较宽,太阳光谱吸收范围窄、光生电子-空穴对易复合、光量子效率低,严重制约其工业化应用。为了拓宽TiO2的光(可见光或太阳光)响应范围,提高光催化性能,常采用金属(Mn、Fe、Ni、Ru、Cu、Ce、Zn和Mo等)和(或)非金属(C、N、S、Cl等)对其进行掺杂改性来提高光催化性能[4-7]。Sharotri等[8]发现Mn和N共掺杂使TiO2的带隙明显变窄(2.4 eV),在波长为660 nm的可见光照射下,对有机磷酸酯类农药喹啉和2-氯苯酚的最大降解率分别为87.5%和91.7%。这主要是因为掺杂的Mn2+取代晶格中Ti4+,形成氧空位,加速相变,混晶效应使共掺杂TiO2展现出良好的催化活性。Tbessi等[9]利用溶胶-凝胶法制备的Mn,Ce共掺杂TiO2光催化降解双氯芬酸(DCF),光催化反应动力学符合准一级Langmuir-Hinshelwood模型,表观速率常数为0.012 min-1,DCF去除率达到94%。在LED可见光照射60 min后,Fe,N共掺杂TiO2分别使酸性橙7的脱色率和矿化度分别达到了90%和83%主要是Fe和N离子成功掺入TiO2晶格中,使其带隙变窄(2.7 eV),该催化剂在处理时间和能量消耗方面均表现出明显优势[10]。同样,Fe,Pr共掺杂TiO2也在去除酸性橙7表现出很高的催化活性[11]。
目前,TiO2制备方法主要有水热法、沉淀法、气相沉积法、微乳液法和微波法等,但这些方法都需多步操作、使用昂贵设备、条件较苛刻且效率低下。然而,超声辅助溶胶-凝胶法是制备共掺杂TiO2的一种十分有效的方法,可以有效地缩短反应时间、控制产物粒径尺寸、促进固体新相的生成、使样品颗粒均匀分布,同时有利于提高元素的掺杂效率[12]。本研究以硝酸铟、硼酸和硝酸铈和分别作为铟、硼和铈源,采用超声辅助的溶胶-凝胶法制备B,In和Ce,In共掺杂TiO2光催化剂,并采用XRD、XPS、UV-Vis DRS、SEM和FT-IR等研究掺杂元素对TiO2结构及催化性能的影响。
1 实 验
1.1 催化剂制备
采用超声辅助溶胶-凝胶法制备B,In共掺杂TiO2光催化剂。具体制备过程见课题组已发表的文章[13-14]。将所制备的催化剂标记为B(5),In(0.1)-TiO2(600)(5,0.1分别为B、In与Ti的原子比率,600为焙烧温度)。用同样方法制备Ce(1),In(1)-TiO2(400)催化剂。文中B,In-TiO2和Ce,In-TiO2所指代内容和上述标记的一致。
1.2 光催化性能测试
在5~7月份晴天进行光催化实验,通过测定罗丹明B(RhB)溶液的光催化降解程度来评价太阳光照条件下催化剂的活性。将50 mg样品加到50 mL(20 mg/L)的RhB溶液中,避光搅拌30 min,以达到吸附-解吸平衡,然后在太阳光照射下进行光催化降解。间隔时间取样,高速离心分离,取上清液,通过紫外-可见分光光度计(SPECORD50,德国耶拿)在554 nm处测定溶液中RhB的浓度。用式(1)计算降解率:
(1)
ln(C0/C)=kt
(2)
式中:η表示降解率;C0、C分别表示降解前,后溶液的浓度;k准一级动力学速率常数(min-1)。
1.3 光催化剂表征
通过X射线衍射仪(XRD)(D/max 2500,日本理学)测定样品晶型结构并计算晶粒尺寸(式(3)),测定条件为:室温,Cu Kα射线(λ=0.15406 nm),扫描范围(2θ=10°~90°)和步长为0.02°;通过扫描电子显微镜(SEM)(S-4800,日本日立)观察共掺杂TiO2样品的微观形貌;通过X射线光电子能谱仪(XPS)(ESCALab250Xi,赛默飞世尔)测定样品表面的元素组成和化学态;采用UV-2450型带积分球的紫外-可见分光光度计测定样品的紫外-可见漫反射光谱,扫描范围200~1 000 nm;利用红外光谱仪(8400S,日本岛津)研究样品的特征官能团。
D=0.89λ/βcosθ
(3)
式中:D为晶粒尺寸,λ为入射线波长,β为衍射峰的半高宽。
2 结果与讨论
2.1 XRD谱
不同催化剂B,In-TiO2和Ce,In-TiO2的XRD谱图和晶格参数如图1和表1所示。由图1可见,两种催化剂的晶相均为单一的锐钛矿相,在2θ=25.36°,37.02°,37.90°,38.60°,48.16°,54.02°,55.22°,62.88°,68.91°,70.35°和75.18°处的衍射峰与锐钛矿相TiO2的(101),(103),(004),(112),(200),(105),(211),(204),(116),(220)和(215)晶面一一对应(PDF No.21-1272,JCPDS)。谱图中没有出现与B、In和Ce元素相关的衍射峰,也未观察到衍射峰移动的现象,这表明掺杂的元素没有形成新的物相或者以其他方式(进入间隙或取代等)均匀地掺杂分散在TiO2中。另外,两种催化剂在2θ=25.36°处的特征衍射峰很尖,说明所制备共掺杂TiO2晶格结构较完整。根据Scherrer公式,选取晶面(101)的衍射峰,计算B,In-TiO2和Ce,In-TiO2催化剂的粒径分别为15.7和10.7 nm。
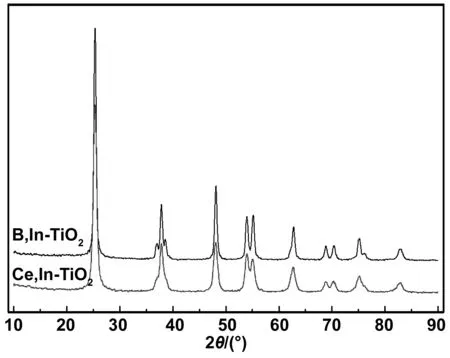
图1 B,In-TiO2和Ce,In-TiO2的XRD图谱Fig 1 XRD patterns of B,In-TiO2 and Ce,In-TiO2 catalysts

表1 催化剂的粒度和晶格参数Table 1 Crystallite size and lattice parameter of all synthesized samples
由于Ce3+(0.103 nm)和In3+(0.081 nm)的离子半径大于Ti4+(0.068 nm),Ce3+和In3+离子几乎不可能以间隙式掺杂方式进入TiO2晶格。如果TiO2晶格中的Ti4+被In3+或Ce3+取代后,晶胞体积增大,衍射峰会向小角度方向移动[15]。从图1可见,Ce,In-TiO2的XRD衍射峰并未出现移动且晶胞体积与标准TiO2几乎一致(见表1)。这表明:Ce3+和In3+未以取代的方式掺入到TiO2晶格之间。因此,Ce3+可能以金属氧化物的形式高度分散在TiO2表面,而In3+可能被化学吸附在TiO2的表面[16-17]。此外,Ce和In元素同时掺入,由于叠加效应,二者能有效抑制晶粒长大,使Ce,In-TiO2的粒径明显小于B,In-TiO2。然而,B3+的离子半径(0.023 nm)远远小于Ti4+(0.068 nm)和O2-(0.132 nm)。TiO2晶格中的Ti4+或O2-被B3+取代时,TiO2的晶胞体积减小,衍射峰向大角度方向移动[18]。实验结果可见,B,In-TiO2的XRD衍射峰并未出现移动,B3+可能以填隙方式进入TiO2的晶格,引起晶格的扭曲,使晶格参数和晶胞体积均减小。
2.2 X射线光电子能谱(XPS)
B,In-TiO2催化剂的表面元素组成及其所处的化学环境如图2的XPS谱所示。从全光谱2(a)可以看出存在Ti2p、O1s、In3d、B1s和C1s,其中C1s峰可能来自于样品暴露在空气中的污染碳或TiO2的前躯体。B1s的标准结合能在B2O3或H3BO3中为193.1 eV(B-O键),TiB2中为187.5 eV(B-Ti键)[19]。从图2(b)可知,B1s的结合能为191.6 eV,与B-O和B-Ti键均无关且大于190.6 eV(B以取代方式掺杂进入TiO2的晶格),这说明B以填隙的方式掺杂到TiO2晶格,B原子可能通过sp2杂化与晶格O原子结合形成B-O-B结构,也可能与O原子和两个Ti原子结合形成Ti-O-B-O-Ti结构[20]。由图2(c)可见,O1s拟合成两个峰,在529.4和530.9 eV分别对应于晶格氧和吸附氧两种化学态氧,其以Ti-O键和-OH形式存在。

图2 B,In-TiO2的XPS谱图(a)全谱,(b)B 1s,(c) O 1s,(d) In 3d,(e) Ti 2pFig 2 XPS spectra of B,In-TiO2 (a) survey spectrum,(b)B 1s,(c) O 1s,(d) In 3d,(e) Ti 2p
在444.4和452.2 eV处出现了In 3d5/2和In 3d3/2光电子峰,如图2(d)所示。这些结合能高于金属In的In 3d5/2(443.8 eV),接近In2O3的In 3d5/2(444.6 eV)[21],这表明除少部分掺杂In3+取代Ti4+形成Ti-O-In结构外,大部分In3+离子以In2O3形式存在于TiO2的表面。由于O离子电负性(3.50)比In离子的电负性(1.78)大,致使Ti-O-In中In 3d结合能高于金属In中In 3d的结合能。图2(e)中,458.2 eV和463.9 eV处的结合能峰属于Ti 2p3/2和Ti 2p1/2光电子峰。ΔEb=Eb(Ti 2p1/2) -Eb(Ti 2p3/2)=5.7 eV,表明样品中的钛处于Ti4+的完全氧化状态,尽管部分Ti4+位点被In3+离子取代,但不影响其氧化状态[22-23]。
In,Ce-TiO2催化剂的表面元素组成及其所处的化学环境如图3的XPS谱所示。从全光谱3(a)可以看出存在Ti2p、O1s、In3d、Ce3d和C1s,其中C1s峰可能来自于样品暴露在空气中的污染碳或TiO2的前驱体。图3(b)中,457.99和463.69 eV处的结合能峰属于Ti2p3/2和Ti2p1/2光电子峰。ΔEb=Eb(Ti 2p1/2) —Eb(Ti 2p3/2)=5.7 eV,表明In,Ce-TiO2中的钛处于Ti4+的完全氧化状态。图3(c)中,结合能为444.06 eV处出现了In3d5/2特征峰,与文献报道的443.8 eV处金属铟的In3d5/2峰相比,排除了金属铟的存在,其接近In2O3的In3d5/2结合能(444.6 eV)[21],这表明In3+离子以In2O3形式存在于TiO2的表面。图3(d)所示为催化剂中Ce3d峰,由于Ce的负载量相对较低,导致其峰信号较弱。Ce的XPS峰受Ce4f和O2p轨道杂化的影响,呈现4对自旋轨道分裂带。v和u分别为Ce的3d5/2和3d3/2自旋-轨道耦合。据文献可知,峰v-u,v″-u″和v‴-u‴分别对应于CeⅣ(3d94f2)O(2p4)、CeⅣ(3d94f1)O(2p5)和CeⅣ(3d94f0)O(2p6)电子态,这6个峰为Ce4+的特征峰;而峰v′-u′对应于CeⅢ(3d94f2)O(2p5)电子态,其为Ce3+的特征峰[24-25]。此外,Ce4+和Ce3+的离子半径分别为0.092和0.103 nm,远远大于Ti4+(0.068 nm),其不可能取代Ti4+进入TiO2晶格。由此可知,Ce以Ce3+/Ce4+氧化物的形式共存于TiO2的表面。O1s拟合成两个峰(图3(e)),在529.21 eV处为TiO2和CeO2体相中的晶格氧,530.66 eV为表面羟基氧,两种化学态氧的存在有利于In,Ce-TiO2催化活性的提高。
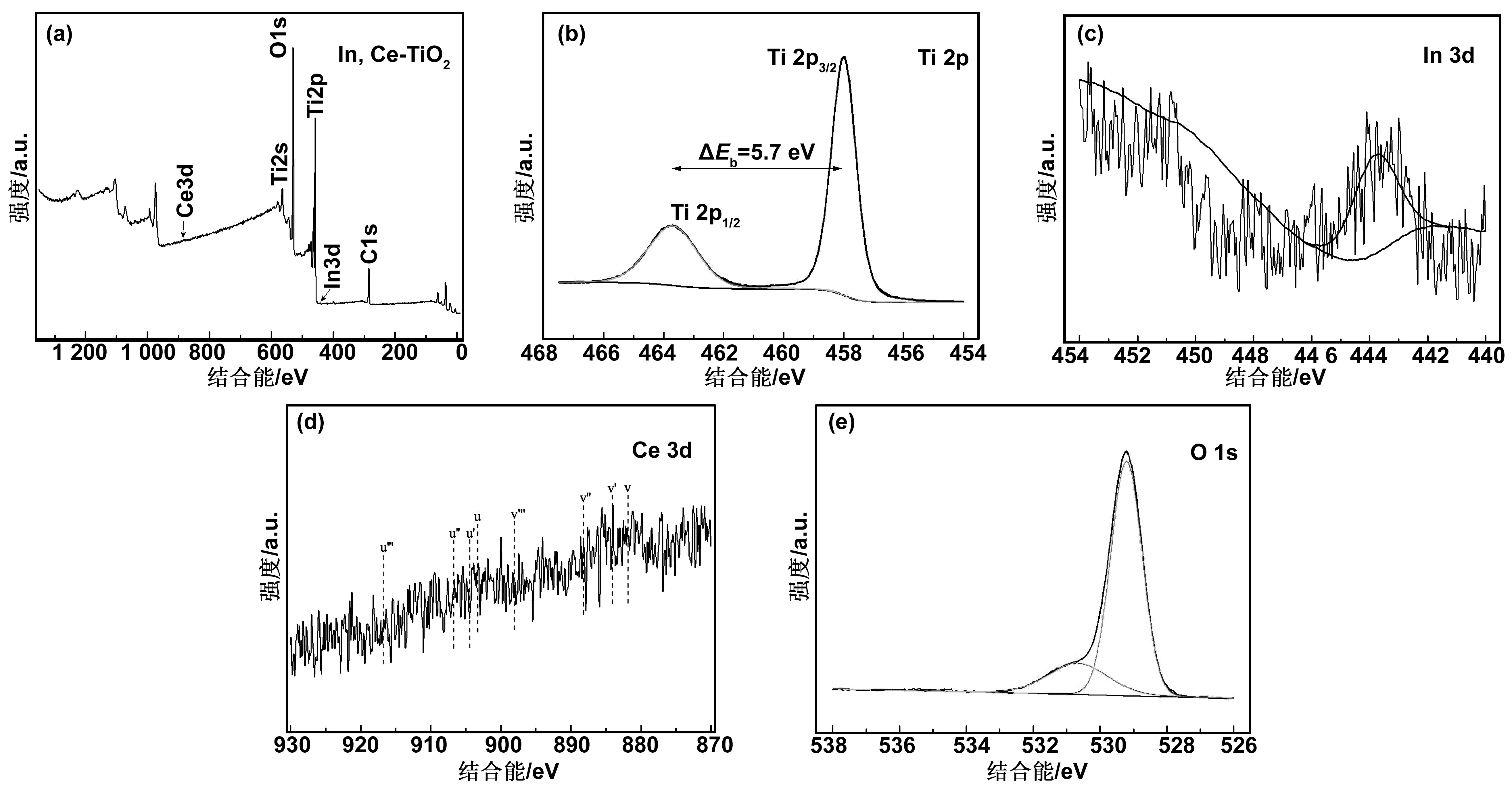
图3 In,Ce -TiO2的XPS谱图(a)全谱,(b) Ti 2p,(c) In 3d,(d) Ce 3d,(e) O 1sFig 3 XPS spectra of In,Ce-TiO2 (a)survey speetrum,(b) Ti 2p,(c) In 3d,(d) Ce 3d,(e) O 1s
2.3 紫外-可见漫反射吸收光谱(DRS)
B,In-TiO2和In,Ce-TiO2的UV-Vis DRS谱如图4所示。由图可见,两种催化剂均在的紫外光区(200~380 nm)有较强的吸收,这是由于价带O2p轨道上的电子被激发跃迁至导带Ti2p轨道的缘故[26]。在波长大于380 nm的可见光区,In,Ce-TiO2(与B,In-TiO2相比)的可见光吸收明显增强,且吸收带出现了一定程度的红移,这是由于In和Ce共掺杂在TiO2带隙中产生了新的能级,使其带隙减小。另外,吸收带的红移表明了In-O-Ti和Ti-O-Ce键的存在。

图4 光催化剂的UV-Vis漫反射吸收光谱图Fig 4 UV-Vis diffuse reflectance spectra of TiO2 photocatalysts
利用Tauc方程(αhv)1/2=A(hv-Eg)计算两种光催化剂的带隙宽度(见图4插图)。与纯TiO2(3.25 eV)[14]相比,B,In-TiO2和In,Ce-TiO2的带隙宽度明显变小,分别为2.96和2.85 eV。对于前者而言,B和In的共掺杂所形成的B-O-B,Ti-O-B和Ti-O-In结构对带隙宽度变窄起了积极作用。对于后者,存在于TiO2表面的Ce的氧化物能与TiO2形成异质结间的相互作用,加之,共掺杂的In和Ce元素对TiO2电子结构的协同作用,使TiO2的吸收边向可见光区红移,带隙变窄[9,27]。
2.4 红外光谱(FT-IR)
样品B,In-TiO2和In,Ce-TiO2的FT-IR谱如图5所示。二者均在3 440和1 640 cm-1处的特征峰分别为羟基的伸缩振动和吸附于其表面的水的弯曲振动。B,In-TiO2催化剂在1 400 cm-1附近的特征峰为Ti-O-B-O-Ti结构中B-O键的伸缩振动,这是B以填隙的方式掺杂到TiO2晶格的结果[18,28],与XPS分析结果一致。在450~700 cm-1处存在B-Ti-O或B-In-Ti的宽吸收峰,这可能是少量In进入TiO2晶格及B取代部分O的缘故。另外,In,Ce-TiO2存在较明显的-OH吸收峰,说明In,Ce共掺杂能更有效的改善TiO2表面形态,增加·OH的数量,其在太阳光照射下有机污染物的降解过程中起到了至关重要的作用。

图5 共掺杂TiO2的FT-IR图谱Fig 5 FT-IR spectra of B,In-TiO2 and In,Ce-TiO2
2.5 扫描电镜(SEM)
由SEM表征的B,In-TiO2和In,Ce-TiO2两种催化剂的微观形貌如图6所示。由SEM图片可见,两种样品的形貌差别不大、均呈球形颗粒、粒径均匀、结构较为致密;B,In共掺杂TiO2出现较多葡萄状堆积,而In,Ce-TiO2类似的堆积较少,主要是由于In3+和Ce4+/Ce3+的存在影响了TiO2颗粒团聚趋势[16]。此外,超声辅助的溶胶-凝胶法制备催化剂过程中,超声波的空化作用所产生的局部高温(5 000 ℃)、高压(50 662.5 kPa),加速水分的蒸发,减少溶胶表面水分子的吸附;加之空化产生的微喷射流冲击界面,使气泡在破灭时变形以及高达100 m/s的细流速度,具有粉碎固体作用,能使已形成的团聚体破碎,释放被包含的水分子,阻止了氢键的形成,达到阻止团聚的目的[29]。

图6 共掺杂TiO2的SEM照片Fig 6 SEM images of B,In-TiO2 and In,Ce-TiO2
2.6 光催化活性
B,In-TiO2和In,Ce-TiO2不同时间光催化降解RhB后的吸收光谱变化如图7(a和b)所示,随着光照时间的延长,RhB逐渐被降解,降解后溶液的最大吸收波长出现了蓝移,这可能是光照降解过程中逐步形成了一系列脱乙基中间体的缘故,也说明RhB的共轭发色团结构易被破坏[30]。RhB主要通过羧基吸附在共掺杂TiO2表面[31],光照产生的活性氧主要攻击发色团和环状结构,其共轭体系被破坏,最终降解率超过99%。

图7 不同光照时间RhB降解的紫外-可见吸收谱图((a)和(b)),降解率和准一级光催化动力学((c)和(d))Fig 7 UV-vis absorption spectra evolution as a function of time for the RhB photodecomposition over B,In-TiO2 and In,Ce-TiO2,photodegradation rate and pseudo-first-order kinetics
图7(c和d)是以准一级动力学方程(式2)拟合的结果,B,In-TiO2和In,Ce-TiO2降解RhB的速率常数分别为0.7907和0.8525 min-1,表明In,Ce共掺杂能更有效的改善TiO2表面形态,增加活性氧的数量。另外,存在于TiO2表面的In2O3和Ce的氧化物使禁带宽度减小,In2O3和CeO2都具有较窄的带隙和强的储氧能力,使得In,Ce-TiO2在太阳光下有较强的催化活性。
2.7 In,X(X=B,Ce)-TiO2光催化机理


图8 光催化降解RhB的可能机理Fig 8 The proposed mechanism of the photodegradation process of RhB
3 结 论
(1) 超声辅助的溶胶-凝胶法制备的B,In-TiO2和In,Ce-TiO2光催化剂,在太阳光照射下对RhB降解率超过了99%。
(2) XRD、XPS、DRS、SEM和FT-IR分析表明:共掺杂能显著改善TiO2表面形态,增加·OH的数量;在B,In-TiO2中,填隙B离子的B2p与O2p轨道杂化形成混合价带,部分In取代TiO2晶格中的Ti,形成Ti-O-In结构,使带隙变窄,光催化活性增强。在In,Ce-TiO2中,Ce的氧化物能与TiO2形成异质结间的相互作用及共掺杂In和Ce元素对TiO2电子结构的协同作用,使TiO2的吸收边红移,带隙变窄。
(3)B,In-TiO2和In,Ce-TiO2降解RhB的速率常数分别为0.7907和0.8525 min-1。
