Theoretical study on the inert C—H arylation and alkylation by metallaphotoredox catalysis*
2022-07-06ZHENGXiaofanZHANGBeibeiLIDeqingCHENBozhen
ZHENG Xiaofan, ZHANG Beibei, LI Deqing, CHEN Bozhen
(School of Chemical Sciences, University of Chinese Academy of Sciences,Beijing 100049, China)
Abstract Metallaphotoredox, including organic photoredox and transition metal catalysis, has emerged as a forceful method to realize the inert C—H bond functionalization to construct C—C or C—heteroatom bonds. In this research, the detailed mechanisms for sp3 C—H arylation and alkylation of tetrahydrofuran (THF) with aryl halide and alkyl halide catalyzed by the triplet excited diaryl ketone and nickel complex have been investigated using density functional theory calculations. The calculations indicate that the whole reaction includes three reaction processes: the formation of THF radical, generation of the product catalyzed by nickel catalysts (nickel catalysis), and regeneration of the Ni catalyst. The THF radical could be produced by the triplet ketone extracting the H atom of THF. For the nickel catalysis, NiL (L=5,5′-dimethyl-2,2′-bipyridine), not Niacac, plays an important role in the arylated reaction. In addition, the Na2CO3 species should be indispensable to the regeneration of NiL. Moreover, the similar results have been obtained for the sp3 C—H alkylation catalyzed by NiL′ (L′=4,4′-diterbutyl-2,2′-bipyridine).
Keywords ketone catalysis; nickel catalyst; DFT; C—H arylation; C—H alkylation
On account of the abundant C—H bonds in nature, the direct modification of nonactive C—H bonds to forge complex molecular structures has been vastly investigated in organic synthesis. The selective functionalization of C—H bonds catalyzed by transition metal has emerged as a key strategy in modern organic chemistry. In recent decades, numerous excellent strategies through transition metal catalysis to achieve challenging C—H bond functionalizations have been reported, such as ruthenium[1], rhodium[2-4], palladium[5], copper[6], iron[7]or nickel[8-10]complexes, wherein the directing groups, stoichiometric additives, strong acids or oxidants[11-14]are generally needed, which might be a deficiency of the synthetic methods.
Over the past four decades, visible light photocatalysis has been tremendously employed in water splitting[15-16], CO2reduction[17], and the exploitation of novel solar cell materials[18]. Nevertheless, until recent years, the application of this catalytic method come to be recognized in organic synthesis[19-20]. In 2007, the Osawa et al.[21]took the lead in applying photocatalysis to the transition metal Pd catalyzed C—C coupling reactions, obtaining significantly increased yield. Later in 2011, Kalyani et al.[22]successfully realized the direct C—H arylation at room temperature by employing the aryldiazonium salts, combining visible-light photoredox catalysis with Pd catalysis.
Inspired by the elegant work of the Osawa and Sanford group, a groundbreaking catalysis platform was emerged at the right moment, termed as metallaphotoredox catalysis, combining photoredox catalysis with transition-metal catalysis. Numerous advances using the metallaphotoredox catalysis were published, exhibiting its unique advantages in C—H functionalization to forge C—C and C—heteroatom bonds[23-25]. Among the plentiful publications, the vast majority of C—H activations were achieved with the utility of precious transition metal catalysts. Concerning the deficiency of these 4 d and 5 d transition metals as being cost-intensive and usually quite toxic[26], the development of powerful strategies relying on earth-abundant and less toxic 3 d transition metal catalysts has been in growing demand. Recent advances have demonstrated a remarkable interest in the exploitation of nickel catalyzed C—H functionalization reactions[27-31]. In terms of publications of recent years, it has to be mentioned that the MacMillan group have developed valuable technologies in the field of direct C(sp3)—H bond alkylation and arylation by using nickel catalyst in conjunction with photoredox catalyst such as Ir[dF(CF3)ppy]2(dtbbpy)PF6[32-34]. However, taking the aforementioned economic factors into consideration, the necessity of probing into novel and alternative photocatalysts for Ir complexes, the widely used species in the photoredox catalysis part, is desirable. Several studies have clarified that carbonyl compounds such as benzaldehyde, acetophenones, quinones and diaryl ketones can be used as efficient photosensitizers and H atom abstractors, in terms of their advantageous photochemical properties as accessible and long-lived triplet states[35-38].
In 2018, Martin and co-workers put forward a dual catalysis platform, combining diaryl ketones for the photoredox catalysis and the nickel catalyst for the transition metal catalysis, which accomplished thesp3C—H bond arylation and alkylation of tetrahydrofuran(THF) with aryl halides and alkyl halides at room temperature, respectively[39]. They found thatsp3C—H arylation and alkylation occur in the o-hydrogen atom of THF. In addition, a higher yield (95%) of targetsp3C—H arylated product was achieved under the presence of the L ligand (L=5,5′-dimethyl-2,2′-bipyridine), while that was 35% under the absence of the L ligand. Furthermore, the yield ofsp3C—H arylated product is only 5% under the absence of Na2CO3. Moreover, for aryl halides containing various substituents, their reactions all can afford good yields.
Martin’s work caught our great interest in the following questions: (ⅰ) Why the o-hydrogen atom of THF is functionalized? (ⅱ) What is the active catalyst in the reaction system? (ⅲ) In the nickel catalysis, which one of the two reactants could firstly combine with the nickel catalyst? (ⅳ) How the nickel catalyst is regenerated? (ⅴ) Why the Na2CO3species is important to the reaction?
To the best of our knowledge, there is no theoretical exploration of the reaction mechanisms reported in the literature. Herein, in order to unveil the above mentioned questions and deeply understand the reaction mechanism, we performed density functional theory (DFT) calculations on several representative reaction systems.
1 Computational methods
All DFT calculations were carried out with the Gaussian 09 program[40]. Geometry optimization calculations were performed for all the stationary points along the reaction pathways using the B3LYP functional[41-42], with a mixed basis set, SDD[43], for nickel and 6-31G(d,p)[44]for the other atoms (named as BS1). Frequency analysis calculations for all of the stationary points were conducted at the same level as for the geometry optimizations to confirm whether they are local minima (no imaginary frequencies) or transition states (only one imaginary frequency). The intrinsic reaction coordinate (IRC)[45-47]has been calculated to verify whether the obtained transition states are connected with the reactants, intermediates or products. Based on the gas-phase optimized structures, the single-point energies were evaluated using the B3LYP functional with a mixed basis set of SDD for nickel and 6-311+G(d,p)[48]for other atoms (denoted as BS2). In order to obtain the solvation energies, M05-2X[49]functional with solvent model density(SMD)[50-51]in THF solution (used in the experiment[39]) in the self-consistent reaction field (SCRF) was employed, in conjunction with a mixed basis set, SDD for nickel and 6-31 G(d)[44]for other atoms (named as BS3). The enthalpies and free energies in THF solution were obtained from the B3LYP/BS1-calculated frequencies. The B3LYP/BS2 free energies with the M05-2X/BS3 solvent corrections at 298.15 K and 1 atm were used in the following discussion.
2 Results and discussions
Herein, we firstly investigated the detailed reaction mechanisms for thesp3C—H arylation of THF with 4-trifluoromethyl bromobenzene in subsection 2.1. Then the effect of substitutents in aryl halides on thesp3C—H arylation reactions were discussed in subsection 2.2, and the detailed mechanisms forsp3C—H alkylation reactions (see reactionBbelow) were investigated in subsection 2.3.
2.1 Mechanisms for sp3 C—H arylation reactions
Based on the speculation by Shen et al.[39], the whole reaction from reactants1and2to product3for thesp3C—H arylation (reactionA1in Fig.1) can be divided into three reaction processes: formation of THF• radical (ketone catalysis), generation of the product catalyzed by nickel catalysts (nickel catalysis), and regeneration of the Ni catalyst. For easy understanding, the above three reaction processes are illustrated in Fig.1a. Herein, we took the C—H arylation of tetrahydrofuran with 4-trifluoromethyl bromobenzene (named as reactionA1.1) as an example and explored the detailed mechanisms for the three reaction processes in turn.
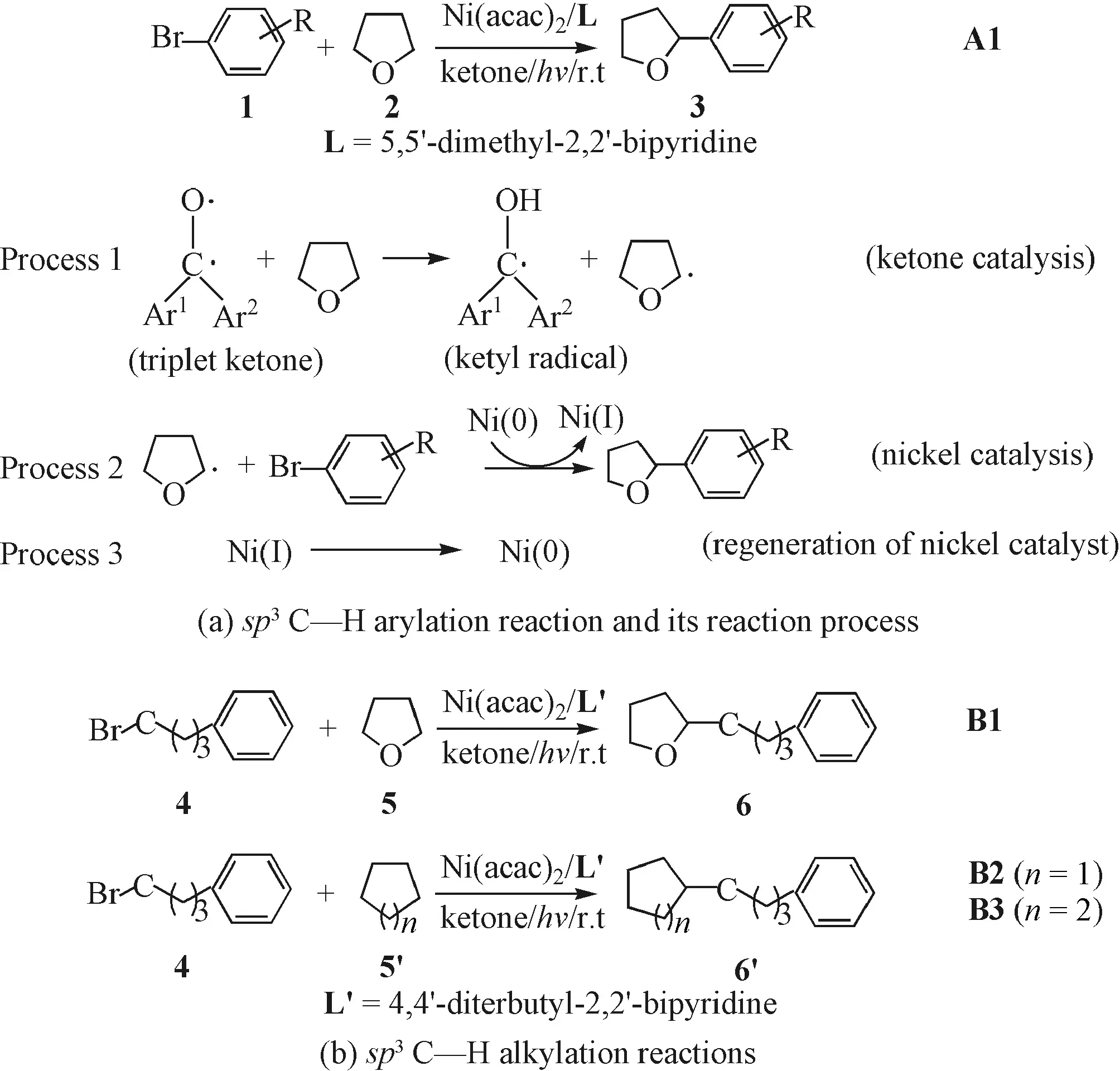
Fig.1 The studied sp3 C—H arylation and alkylation reactions
2.1.1 Formation mechanisms of THF• radicals

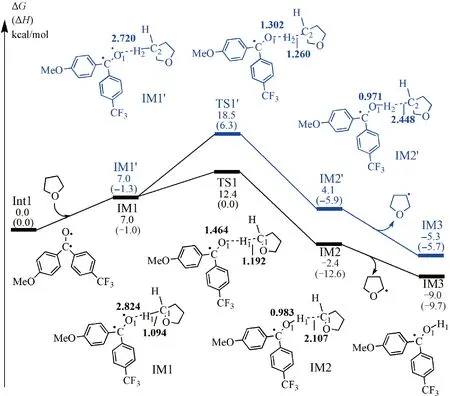
The values were calculated in the solvent of tetrahydrofuran (THF) at 298 K. All bond lengths are given in Å.
2.1.2 Ni-catalyzed reaction processes
ThedeterminationofactiveNicatalysts
In order to determine the composition and structure of Ni catalysts, we optimized the structures of several nickel complexes, such as Ni(acac)2, Niacac, NiL2, and NiL (L=5,5′-dimethyl-2,2′-bipyridine). The calculated results show that these species all correspond to the minima in the potential energy surfaces and their electronic ground states are all in the triplet state. The calculated structures are shown in Fig.S1. For Ni(acac)2, as a nickel resource in the reaction system, its singlet state is energetically 23.4 kcal/mol higher than its triplet state (Table S2). Therefore, only the triplet Ni(acac)2was considered to take part in the reaction processes. Furthermore, considering that the tetracoordinated Ni(acac)2and NiL2should be somewhat difficult to cooperate with reactants due to the steric hindrance, we turned our attention to the other two nickel species (Niacac and NiL). Niacac should be generated from the dissociation of Ni(acac)2, and thus the reaction free energy of the dissociation process was calculated. The value is predicted to be 13.2 kcal/mol, implying that this process is unfavorable thermodynamically. NiL should be formed from the ligand substitution between Ni(acac)2and L. Our calculations indicate that the reaction free energy for this ligand-exchange reaction is -8.2 kcal/mol (Table S3), demonstrating that the process is feasible from a thermodynamic point of view. Therefore, the NiL species may play an important role in the followedsp3C—H arylated product formation. Herein, we calculated the detailed mechanisms of reactions between aryl halide andTHF• catalyzed respectively by NiL (1Cat) and Niacac (2Cat) in the triplet state to figure out the catalytic activities of both the Ni catalysts, although the formation of NiL has advantage over that of Niacac thermodynamically.
20. The ring from my finger: The young woman s offering is the last of her valuable possessions. It is a more symbolic piece of jewelry than a necklace. Rings are used to plight troths and represent formal unions, especially marriages.
Theformationofsp3C—Harylatedproductcatalyzedby1Catand2Cat
The reaction pathways catalyzed by1Catand2Catwere calculated, respectively, and the results are shown in Figs.3 and S2. As can be seen in Fig.3, there are two pathways (Path a and Path b) from the reactants to an intermediate1-IM3for the C—H arylated reaction catalyzed by1Cat. Along Path a,1Catfirstly cooperates with aryl halide, giving rise to a precomplex1-IM1a, and the reaction free energy for this step is 1.0 kcal/mol. Then, the oxidative addition of aryl halide to the nickel center leads to a tetrahedron type Ni(Ⅱ) intermediate1-IM2avia a transition state1-TS1a. Although the free energy of1-IM1ais 1.0 kcal/mol higher than that of the reactants, the followed reaction step (from1-IM1ato1-IM2a) releases the free energy of 52.1 kcal/mol, which could promote the reaction to proceed. It is noted that1-TS1ahas 1.1 kcal/mol lower in free energy than1-IM1a, however,1-TS1ais 1.7 kcal/mol higher than1-IM1ain gas-phase, which might mean the interaction of solvent with this transition state is stronger than that with1-IM1a. Therefore, the oxidative addition of aryl halide to1Catshould take place quite easily both thermodynamically and kinetically. Subsequent coordination of THF• (generated in the triplet ketone catalysis discussed above) to Ni(Ⅱ) intermediate1-IM2ahas only a free energy barrier of 11.4 kcal/mol, leading to a Ni(Ⅲ) intermediate1-IM3with a free energy of -52.2 kcal/mol relative to the reactants. Therefore, Path a is feasible both kinetically and thermodynamically and should play an important role in the formation of arylated product. Along Path b (Fig.3), the NiL firstly cooperates with THF• to form a T-shaped Ni(Ⅰ) intermediate1-IM1b, with a large driving force (ΔG=-32.4 kcal/mol), and no free energy barriers exist in this step (Table S4), demonstrating that this reaction step is favorable both thermodynamically and kinetically. The subsequent oxidative addition of aryl halide to the nickel center of1-IM1boccurs via a transition state1-TS1b, leading to the same intermediate1-IM3as Path a. Although this reaction step undergoes a 24.6 kcal/mol free energy barrier, the free energy released in the previous step is enough to overcome this barrier. It seems that Path b should also play a role in the reaction. However, taking the reaction conditions[39]into consideration, we must mention that the amount of THF• should be far less than that of aryl halide. Therefore, the initial reaction could proceed along Path a, namely, the NiL first cooperates with the aryl halide rather than THF•. Thus, our further discussion will mainly focus on Path a.
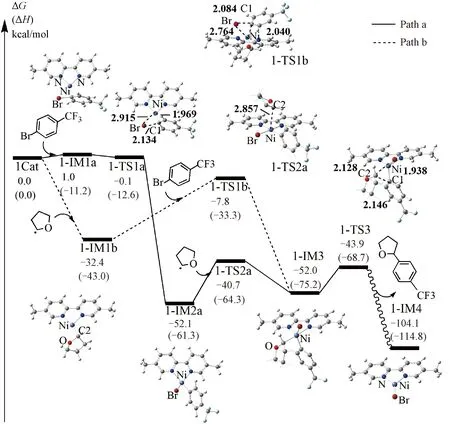
The values were calculated in the solvent of THF at 298 K. Enthalpies are shown in parentheses. All bond lengths are given in Å. Several H atoms have been omitted for clarity.
The followed reaction from1-IM3is the reductive elimination, generating the targeted C—H arylation product and [BrNi(Ⅰ)L]0intermediate1-IM4through a transition state1-TS3with a free energy barrier of 8.1 kcal/mol, and the reaction free energy for this step is predicted to be -52.1 kcal/mol, manifesting that this step occurs easily both kinetically and thermodynamically. In summary, the formation of thesp3C—H arylated product catalyzed by1Catincludes three reaction steps, namely, the oxidative addition of1Catand aryl bromide, the cooperation of THF• to Ni(Ⅱ) intermediate, and the reductive elimination of Ni(Ⅲ) to generate product, and the second one is the rate determined step. The overall free energy barrier (corresponding to the free energy barrier for the rate-limiting step) and overall reaction free energy are 11.4 and -104.1 kcal/mol, respectively, indicating that the1Catcatalyzed C—H arylated reaction is favorable from both kinetic and thermodynamic points of view. It should be mentioned that the experiments were at room temperature and it is noted that the reaction at room temperature usually has a free energy barrier near 20 kcal/mol. However, our calculated free energy barriers are around 12 kcal/mol (see Figs.2 and 3). The mismatch between them might derive from the several possible causes. The first one is that the formation of the active catalyst [1Cat(NiL)] might have higher free energy barriers, that is, the activation of the catalyst may require the room temperature condition. The second one is that the visible light induced reaction might also occur below room temperature. The third one may be that the free energy barrier is underestimated due to the computational method.
The reaction pathway for the2Catcatalyzed C—H arylation is similar to that catalyzed by1Cat(see Fig.S2). The overall free energy barrier and reaction free energy are calculated to be 21.8 and -105.7 kcal/mol, respectively, indicating that2Catalso has catalytic activities for thesp3C—H arylation. However, considering that the formation of2Cathas disadvantage over that of1Cat(see above),1Catshould be the active catalyst for thesp3C—H arylation. In addition, by comparing the reaction pathways catalyzed by1Catand2Cat, it could also be found that the energy barriers for the three key procedures (the oxidative addition of nickel catalyst and aryl halide, the cooperation of THF• to the Ni(Ⅱ) intermediate, and the reductive elimination of Ni(Ⅲ) intermediate to release product) catalyzed by1Cat(1.0, 11.4, 8.1 kcal/mol, Fig.3) are lower than that by2Cat(4.4, 21.8, 8.3 kcal/mol, Fig.S2). Therefore,1Catshould play more important role in the reaction than2Catfrom a kinetic point of view.
In order to give the insight into catalytic activities of both1Catand2Cat, we compare the charge distributions in the key structures along the reaction pathways catalyzed by1Catand2Cat. The results can be seen in Table 1. It is evident that in the structure of1Cat, the ligand L possesses stronger electron-absorbing property than acac, which contributes to the better electropositivity of nickel in1Cat(0.594 e) than that in2Cat(0.554 e). Furthermore, the1Catmoiety (0.268 and 0.480 e) has more positive charges than the2Catmoiety (0.136 and 0.107 e) inn-IM1andn-TS1, showing more electrons move to the aryl halide reactant catalyzed by1Catthan by2Cat. Therefore, the oxidative addition of aryl halide to1Catis more favorable than to2Cat. This may explain the higher energy barrier in the oxidative addition of the aryl halide with2Catthan with1Cat. For the cooperation of THF• to the Ni(Ⅱ) intermediate, because C(sp3)• (the carbon center of THF•) and Ni center for bothn-TS2s all have positive charges (0.100 and 0.095 e for C(sp3)•, 0.392 and 0.447 e for Ni center), C(sp3)• might be more feasible to cooperate with less positive Ni center in1-TS2, which is also consistent with the fact that the distance between the Ni and C2 atoms in1-TS2(2.857 Å) is much shorter than that in2-TS2(3.308 Å). Thus, the reaction step fromn-IM2ton-IM3catalyzed by1Cathas a lower energy barrier than that by2Cat. To sum up, the calculated results indicate that both1Catand2Cathave catalytic activities for C—H arylation reaction but the former is advantageous to be formed and more active than the latter, just explaining the experimental fact that the high yield (95%) of targetsp3C—H arylated product is achieved under the presence of the L (5,5′-dimethyl-2,2′-bipyridine) ligand, while that is lower (35%) under the absence of the L ligand[39].
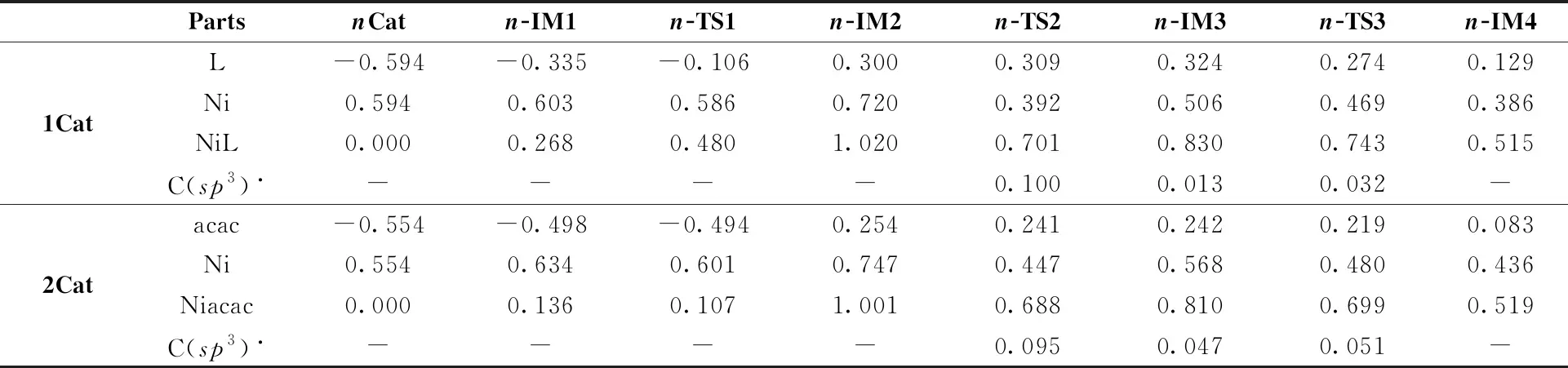
Table 1 The charge distributions of the stationary points along the pathways of sp3 C—H arylated reactions catalyzed by 1Cat and 2Cat
2.1.3 Regeneration of Ni catalyst
To achieve catalytic cycling, the regeneration of Ni catalysts is crucial. There are three possible pathways for the regeneration of the Ni catalyst. We performed calculations for them in detail and the results are shown in Figs.4, S3 and Tables S5, S6. The first pathway includes three reaction steps (Fig.4): the reaction of ketyl radical (IM3in Fig.2) with Na2CO3producing the doublet ketone anion and [HNa2CO3]+cation (Step 1), the single electron transfer (SET) from the doublet ketone anion to1-IM4([BrNi(Ⅰ)L]0) generating the singlet ketone (the ground state) and the [BrNi(Ⅰ)L]-anion (Step 2), and the reaction of [BrNi(Ⅰ)L]-anion and [HNa2CO3]+cation releasing the1Cat(Step 3). It is noted that Step 1 is the acid-base reaction, while Step 3 is the reaction between positive and negative ions. Both reactions should occur easily. The free energy barrier of Step 2 was calculated based on Marcus theory[52-54], and the value is predicted to be 11.1 kcal/mol (Table S5), demonstrating that the process is favorable kinetically. The overall reaction free energy for the regeneration of1Catis predicted to be -23.0 kcal/mol. Therefore, the first pathway should be feasible from both kinetic and thermodynamic points of view. It is worth mentioning that Na2CO3is a key species in this pathway.
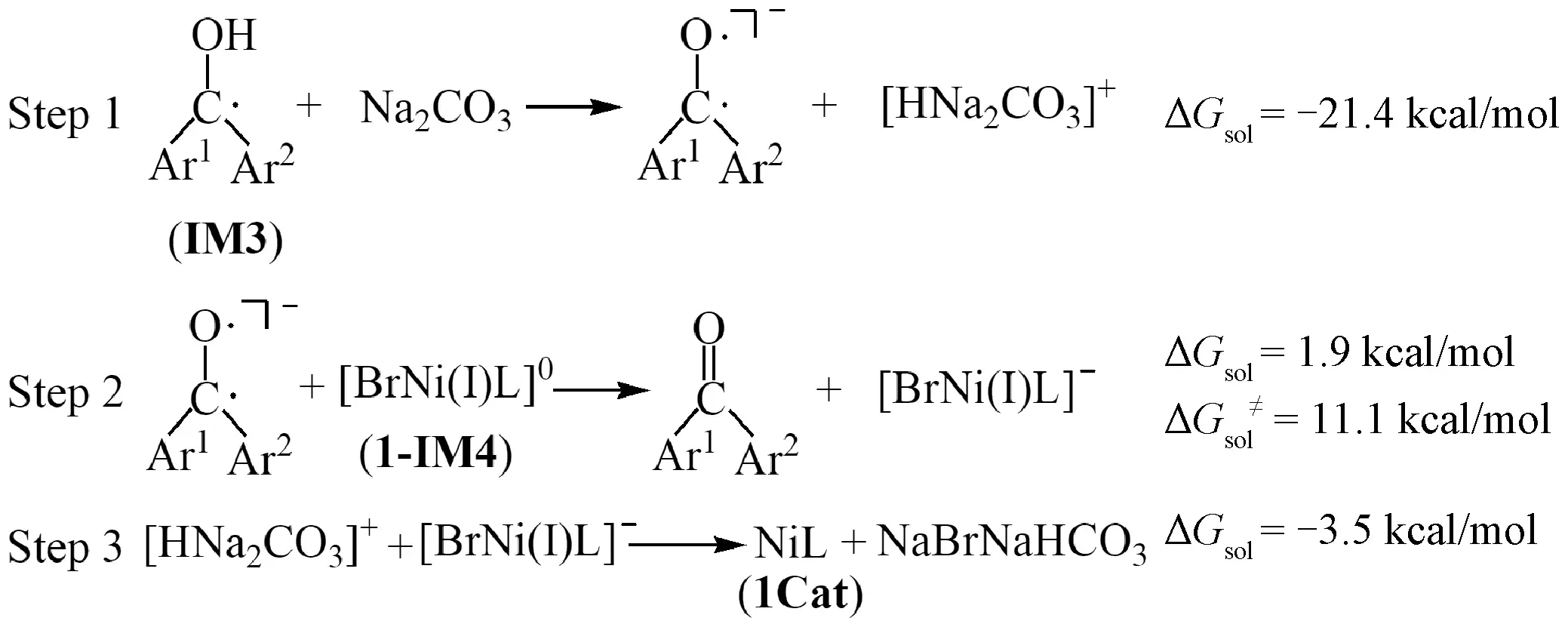
Fig.4 Three reaction processes for regeneration of nickel catalyst (NiL)
The second pathway for the regeneration of the Ni catalyst is the reaction of ketyl radical with [BrNi(Ⅰ)L]0, which was speculated by Martin group[39]. In order to figure out its feasibility, we investigated this pathway in detail and the results are shown in Table S6. Firstly, the reaction of [BrNi(Ⅰ)L]0with ketyl radical leads to the formation of triplet [HBrNiL]0and singlet ketone, and the reaction free energy is -2.5 kcal/mol. Then the triplet [HBrNiL]0dissociates into the triplet NiL and HBr, and the calculated reaction free energy of this process is 48.5 kcal/mol. Obviously, this process is unfavorable thermodynamically. However, the produced HBr can easily react with Na2CO3existed in the reaction system, releasing the free energy of 46.8 kcal/mol, which could promote the regeneration of1Cat. Our results support the speculation by Shen et al.[39]on the one hand, and clearly illustrate the importance of Na2CO3in the regeneration of the Ni catalyst on the other hand.
The third possible pathway is that the ketyl radical may abstract the Br radical in the Ni(Ⅲ) intermediate1-IM3to realize the regeneration of1Cat. Unfortunately, the free energy barrier has at least 34.4 kcal/mol with respect to1-IM3plus the ketyl radical (Fig.S3). This might be a hint that the ketyl radical abstracting Br radical is impossible.
To sum up, our calculations indicate that there are two reaction pathways for the regeneration of the NiL catalyst in the reaction system, and the Na2CO3species plays a key role in both the pathways. The calculated results give a perfect explanation of the experimental facts that the yield ofsp3C—H arylated product is only 5% under the absence of Na2CO3[39].
2.2 Substituent effect of aryl halides on the sp3 C—H arylation
In order to figure out the substituent effect of aryl halide onsp3C—H arylation, the reaction mechanisms between THF and reactant1(see Fig.1) with the different properties catalyzed by1Cat(reactionsA1.2-A3in Table 2) were investigated in detail and the relative free energies of all the stationary points to the corresponding reactants along the reaction pathways are shown in Table 2. For the sake of convenience, reactionA1.1and the experimental yields of target products are also shown in Table 2. It should be noted that reactant1in reactionsA1.2andA1.4was designed by us for later discussion. Considering that these reactions all require THF• (the formation of THF• was discussed in 3.1.1), herein, we mainly focus on the nickel catalysis (Process 2 in Fig.1).
It should be mentioned that the free energy barriers for the eleven reactions are different (see Table 2). To shed light on the better performance of the reactants bearing the electron-withdrawing substituents rather than the electron-donating substituents, we mainly compare the free energy barriers along the potential energy surface for reactionsA1.1-A1.3andA1.6-A1.8with the para-positioned substituents in aryl bromides. The former four reactions contain the electron-withdrawing substituents, while the latter two reactions have electron-donating substituents. In the first oxidative addition, it can be found that for those reactants possessing electron-withdrawing substituents (A1.1,A1.2,A1.3,A1.6), the free energies of1-TS1aare all lower than that of1-IM1a, indicating that these1-TS1as may disappear in the reaction path calculations of the higher-level theory. Therefore, the cooperation of these aryl bromides with1Catmay proceed without any free energy barriers. On the contrary, for those reactants with electron-donating substituents (A1.7,A1.8), the free energies of1-TS1aare higher than that of1-IM1a, being 2.0 and 2.7 kcal/mol, respectively. Then in the following oxidative addition of THF•, the electron-withdrawing group owners (A1.1,A1.2,A1.3,A1.6) undergo somewhat lower free energy barriers (11.4, 15.9, 18.6, and 13.9 kcal/mol, respectively) than the electron-donating group owners (20.1 and 20.0 kcal/mol inA1.7,A1.8separately). The tendency in the reductive elimination is similar to that in the coordination of Ni(Ⅱ) intermediate with THF•. In summary, there is an overwhelming preference for electron-withdrawing substituents over electron-donating substituents, which explains that the yields of the former are higher than that of the latter experimentally. Moreover, it is worth mentioning that the —CN group in reactant1(reactionA1.6) is thermodynamically more favorable to cooperate with nickel center than the C1—Br bond (Fig.S4). This fact may be the reason that reactionA1.6has lower yields than reactionsA1.1andA1.3.
To unveil the effect of the substituent position in aryl halide on the reaction, we designed the meta-positioned ester group of aryl halide (reactionA1.4in Table 2). It can be found from Table 2 that reactionsA1.4andA1.5(ortho-positioned ester group of aryl halide) also have lower free energy barriers, similar to the case of para-positioned one (A1.3). These facts indicate that the substituent position has less effect on the reaction. In other words, the yield of the C—H aryl product might be satisfactory if aryl halides with the meta-positioned ester group is taken as reactant1.
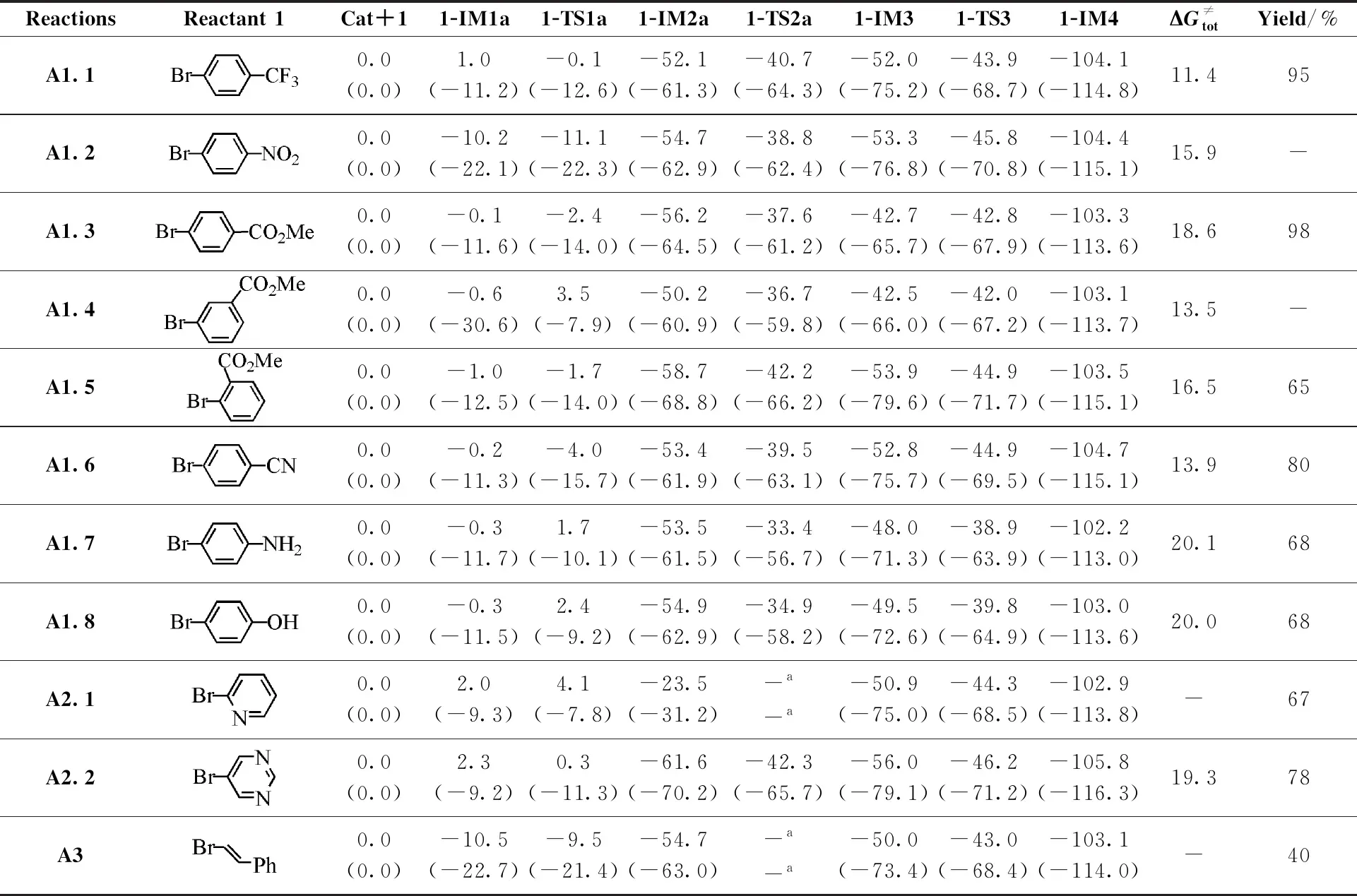
Table 2 The relative free energies of the stationary points along the pathways of sp3 C—H arylated reactions between THF and reactant 1 with various substituents in THF solvent
As shown in Table 2, reactionsA2.1andA2.2with heteroaryl bromide reactants and reactionA3with vinyl bromide reactants all have similar reaction pathways to Path a in reactionA1.1. Also, the three reactions all have low free energy barriers, which demonstrates that they are feasible for thesp3C—H arylation reactions. Based on our calculations, the overall free energy barriers for all the eleven reactions are smaller than 20.1 kcal/mol, and the reaction free energies are between -102.1 and -105.8 kcal/mol. Therefore, all the reactions should occur from both kinetic and thermodynamic points of view. The calculation results are consistent with experiment[39].
2.3 Mechanisms for sp3 C—H alkylation reactions
For thesp3C—H alkylation, we selected reactionB1(see Fig.1(b)) as an example and calculated its detailed mechanism. Since the formation of Niacac is unfavorable (see above), the NiL′ (L′ = 4,4′-diterbutyl-2,2′-bipyridine) was used as the catalyst (named as3Cat). Considering the formation of THF• is the same as that of thesp3C—H arylation reactions (discussed in 3.1.1), herein, we mainly focus on the3Catcatalyzed reaction processes for alkylation reactions. The computed free energy profile for the NiL′ catalyzedsp3C—H alkylation reaction is shown in Fig.5.
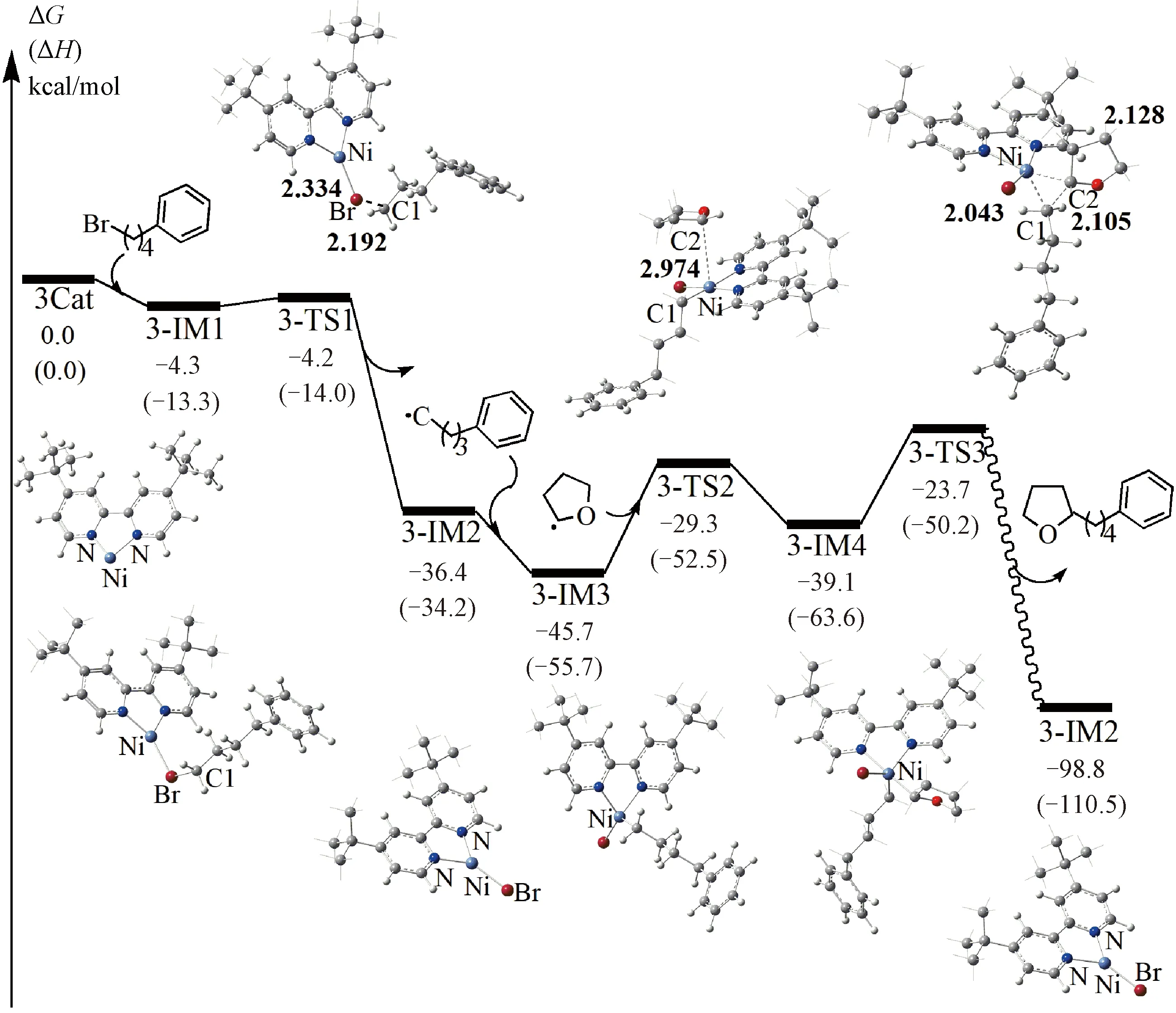
Enthalpies are given in parentheses. The values were calculated in the solvent of THF at 298 K. Bond lengths are given in Å. Several H atoms have been omitted for clarity.
As shown in Fig.5, notably, unlike the direct oxidative addition of aryl bromide to the NiL to afford Ni(Ⅱ) intermediate in thesp3C—H arylation, here in the alkylation pathway, the generation of Ni(Ⅱ) complex requires two steps. First, starting from3-IM1, the cleavage of the C1—Br bond via a transition state3-TS1generates Ni(Ⅰ) intermediate3-IM2and phenylbutyl radical. This process only requires a low free energy barrier (0.1 kcal/mol) and the reaction free energy is -36.4 kcal/mol, which means it is kinetically and thermodynamically accessible. Then the phenylbutyl radical coordinates with the Ni(Ⅰ) intermediate to form the tetrahedron type Ni(Ⅱ) complex3-IM3, with a reaction free energy of -9.3 kcal/mol. Subsequently, the carbon centered radical THF• adds to the3-IM3via a free energy barrier of 16.4 kcal/mol, giving rise to the Ni(Ⅲ) intermediate3-IM4, and the reaction free energy of this step is 6.6 kcal/mol. Then the reductive elimination occurs via a transition state3-TS3, which requires a free energy barrier of 15.4 kcal/mol, producing ansp3C—H alkylated product and releasing a free energy of 59.7 kcal/mol. Similar to the case of1Cat, the regeneration of3Catalso includes three steps (see Fig.S5), and should be favorable thermodynamically and kinetically. We will not discuss it in detail here.
The reaction pathways of the alkyl halide with the cyclopentane and with cyclohexane catalyzed by3Cat(NiL′), named as reactionsB2andB3, respectively, were also investigated. Since one reactant (alkyl halide) of these two reactions are the same as reactionB1, only the ketone catalyzed HAT process and the Ni(Ⅱ)→Ni(Ⅲ)→Ni(Ⅰ) transformation in the nickel catalysis (namely, the cooperation of carbon centered radical with Ni(Ⅱ) intermediate, and the eliminated reduction of Ni(Ⅲ) intermediate to release Ni(Ⅰ) intermediate) need to be explored. The calculated results are given in Fig.S6. It can be found from Fig.S6(a) that, in the ketone catalysis, the generations of cyclopentane radical and cyclohexane radical require the free energy barriers of 15.0 and 14.3 kcal/mol, respectively. Although their free energy barriers are 2.6 and 1.9 kcal/mol higher than that of the THF• radical, they could also occur. Thus, we speculate that both cyclopentane and cyclohexane may be suitable for thesp3C—H arylation reactions. As for the nickel catalysis, it is noted that the two free energy barriers in the Ni(Ⅱ)→Ni(Ⅲ)→Ni(Ⅰ) transformation for reactionB2(B3) are 22.2 (20.7) and 16.3 (15.1) kcal/mol, respectively. These two energy barriers are higher than or similar to those of reactionB1, respectively, as shown in Fig.S6(b). These results indicate that reactionsB2andB3are less favorable than reactionB1in the kinetic point of view, which is consistent with the experimental results (the yield of the former two are 41% and 51%, respectively, and the latter is 86%)[39].
In addition, comparing the reaction pathways of both the C—H alkylation and arylation, we could speculate that the lower yield of the former may also result from the higher free energy barriers in the ketone catalysis and Ni(Ⅱ)→Ni(Ⅲ)→Ni(Ⅰ) transformation processes in the nickel catalysis.
3 Conclusion
Focusing on the reactions of metallaphotoredox catalyzedsp3C—H arylation and alkylation, we performed density function theory calculations for the detailed mechanisms of THF reacting with aryl halides bearing different substituents and alkyl halide and the whole mechanisms are shown in Fig.6. For thesp3C—H arylation, the calculations support that the whole reaction consists of the three reaction processes: the formation of THFradical (ketone catalysis), generation of the product catalyzed by nickel catalysts (nickel catalysis), and regeneration of the Ni catalyst, just as the experimental researchers speculated. The THFradical could be produced by the triplet ketone extracting the H atom of THF with a free energy barrier of 12.4 kcal/mol. In the nickel catalysis, NiL (L=5,5′-dimethyl-2,2′-bipyridine), not Niacac, is predicted to be the active catalyst, and there exist two reaction pathways. One is the cooperation of aryl halide with nickel center firstly, the other is that nickel center firstly combines with THFradical. The former should play a more important role in the reaction. Those reactions for aryl halides containing electron-donating groups have higher free energy barriers than those containing electron-withdrawing groups, but the latter reactions are also feasible kinetically. For the regeneration of the nickel catalyst, there are two potential reaction pathways and the Na2CO3species should play a key role in both the pathways based on our calculations, which gives a perfect explanation of the experimental facts that the yield ofsp3C—H arylated product is only 5% under the absence of Na2CO3.
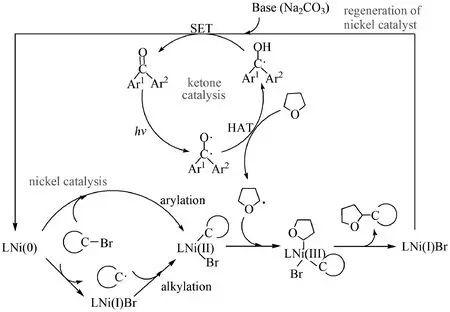
Fig.6 Predicted reaction mechanisms for the sp3 C—H functionalization
For thesp3C—H alkylation, the reaction mechanisms of THF, cyclopentane, and cyclohexane with alky halide were investigated. Our calculations demonstrate that the three reactions have similar mechanisms to the arylation reactions. The formation of the cyclopentane and cyclohexane radicals have only 2.6 and 1.9 kcal/mol higher free energy barriers than that of the THFradical, and both cyclopentane and cyclohexane may be suitable for thesp3C—H arylations. We expect that the present work could provide important information for experimental workers to design thesp3C—H functionalized reaction system.
