QuEChERS-超高效液相色谱-质谱联用法测定熊胆粉中9种苯二类药物残留量
2022-07-01王小乔许晓辉张虹艳吴福祥潘秀丽
王小乔,许晓辉*,张虹艳,冯 翀, 2,吴福祥,潘秀丽,李 赟
(1.兰州市食品药品检验检测研究院,兰州 730050;2.兰州理工大学生命科学与工程学院,兰州 730050)


1 材料与方法
1.1 仪器 Agilent 1290 UPLC/6460C超高效液相色谱-串联三重四极杆质谱联用仪,配有电喷雾离子源(美国安捷伦科技有限公司);Vortex-genie2涡旋混合器(美国Scientific);H1850离心机(湖南湘仪);MS105DU分析天平(梅特勒-托利多);Milli-Q超纯水系统(美国Millipore公司);SBL-10DT超声波清洗器(宁波新芝生物科技股份有限公司,功率为300 W);GENO2010组织研磨仪(美国SPEX)。
1.2 试剂 甲醇、乙腈、乙酸乙酯(色谱纯,德国Merck公司);甲酸、乙酸铵(色谱纯,东京化成工业株式会社); EMR-Lipid dSPE 净化管(美国安捷伦科技有限公司);硫酸钠、氯化钠(分析纯,国药集团化学试剂有限公司)。对照品:马来酸咪达唑仑(批号:171250-201002;纯度:99.9%)、艾司唑仑(批号:171219-201003;纯度:99.7%)、奥沙西泮(批号:171229-201505;纯度:99.8%)、硝西泮(批号:171217-201403;纯度:99.9%)、劳拉西泮(批号:171253-201102;纯度:99.2%)、阿普唑仑(批号:171218-201305;纯度:99.5%)、氯硝西泮(批号:171227-201404;纯度:99.9%)、三唑仑(批号:171230-201203;纯度:99.6%)、地西泮(批号:171225-200903;纯度:99.9%)均购自中国食品药品检定研究院;熊胆粉样品购自中药材市场。
1.3 溶液制备
1.3.1 EDTA-Mcllvaine缓冲液 称取柠檬酸8.4 g,Na2EDTA 19.5 g,磷酸氢二钠7.1 g,溶于650 mL水中,超声溶解。
1.3.3 供试品溶液的制备 提取:取熊胆粉样品适量,研细,混匀;精密称取熊胆粉约1 g置于50 mL离心管中,加入3 mL EDTA-Mcllvaine缓冲液,加入1粒陶瓷均质子,振荡摇匀,再加入乙腈10.00 mL,在组织研磨仪上振荡涡旋5 min,在300 W功率下超声提取5 min,加入含4 g 硫酸钠和1 g 氯化钠的萃取盐包,再涡旋2 min,在4 ℃下以4000 r/min速率离心15 min,取上清液待净化。
净化:精密移取上述上清液2.00 mL于EMR-Lipid dSPE净化管中,涡旋1 min,在4 ℃下以4000 r/min离心15 min,取上清液过0.22 μm滤膜,供液相色谱-质谱联用仪测定。
1.4 色谱-质谱条件
1.4.1 色谱条件 ACQUITY BEH C18色谱柱(1.7 μm,2.1×100 mm);流动相:以含0.1 %甲酸-2 mmol/L乙酸铵的水溶液为流动相A,以含0.1%甲酸的乙腈溶液为流动相B,梯度洗脱程序:0~3.0 min,10%~30%B;3.0~5.0 min,30%~90%B;5.0~7.0 min,90%B;7.0~8.0 min,90%~10%B;8.0~9.0 min,10%B。柱温:35 ℃;进样量: 2 μL;流速:0.3 mL/min;分析时间:9 min。
1.4.2 质谱条件 电离源:喷射流电喷雾离子源(AJS ESI);离子源模式:正离子模式;采集方式:多反应监测模式(MRM);干燥气:N2;干燥气流量:8 L/min;干燥气温度:325 ℃;雾化气压力:45 psi;毛细管电压:4000 V;鞘流气流量:12 L/min;鞘流气温度:350 ℃,相关质谱采集参数见表1。
2 结果与分析
2.1 前处理条件优化
2.1.1 提取溶剂的选择 本实验分别选择甲醇、乙腈、乙酸乙酯为提取溶剂,以相同水平浓度的9种目标分析物加标对应回收率为依据,按照1.3.3和1.4项下条件进行前处理和上机分析,考察提取溶剂的提取效果。结果表明,以甲醇为提取溶剂时,回收率处于15%~56%之间,且大多数目标分析物的回收率小于40%;以乙酸乙酯为提取溶剂时,回收率处于51%~180%之间,且除硝西泮之外,其余目标分析物回收率普遍偏高;以乙腈为提取溶剂时,目标分析物的回收率整体较好,这可能是乙腈对目标分析物具有较好的溶解性,同时能很好地沉淀蛋白质,去除杂质比较充分,整体上降低了基质的干扰(图1)。因此,最终选择乙腈为提取溶剂。

图1 不同提取溶剂对9种苯二类药物提取效率的影响Fig 1 Influence of different extraction solvents on theextraction efficiency of nine benzodiazepine drugs
2.1.2 净化方法的选择 由于熊胆粉样品基质中含有较多熊去氧胆酸、鹅去氧胆酸、胆酸、去氧胆酸及少量脂肪和蛋白质,这些类别的杂质会在提取过程中进入提取液,在分析目标化合物时会产生基质效应,对检测结果造成干扰,这些杂质大部分是脂溶性的,因此需要采用高脂类净化方式来除杂,而增强型脂质去除分散固相萃取净化管(EMR-Lipid dSPE)是专门为多脂样品中脂质性杂质的选择性净化而设计,因此,本实验选择EMR-Lipid dSPE方式进行净化。
2.2 色谱质谱条件优化


表1 9种苯二类药物检测离子对及相关参数Tab 1 Ion pairs and related parameters for nine benzodiazepine drugs
2.3 基质效应 本实验通过取空白样品,按照前处理方法制备成空白样品基质溶液,再分别用空白样品基质溶液与纯溶剂配制成相同浓度的对照品溶液,以两者同一药物质谱响应值比值的百分数来评价基质效应(Matrix Effect,ME),若ME大于100%,则存在基质增强效应;若ME小于100%,则存在基质抑制效应;若ME接近100%,则不存在基质效应,本实验设计了两个浓度级别10 ng/mL和30 ng/mL,结果发现,9种化合物在10 ng/mL的条件下,除劳拉西泮存在基质效应增强效应外,其余分析物存在一定程度的基质抑制效应,其中基质抑制效应最强的是咪达唑仑和硝西泮;在30 ng/mL的浓度下,艾司唑仑、奥沙西泮、阿普唑仑存在一定程度的基质增强效应,其余待测物存在不同程度的基质抑制效应。虽然各待测物存在不同程度的基质效应,但不影响检测结果(图2)。
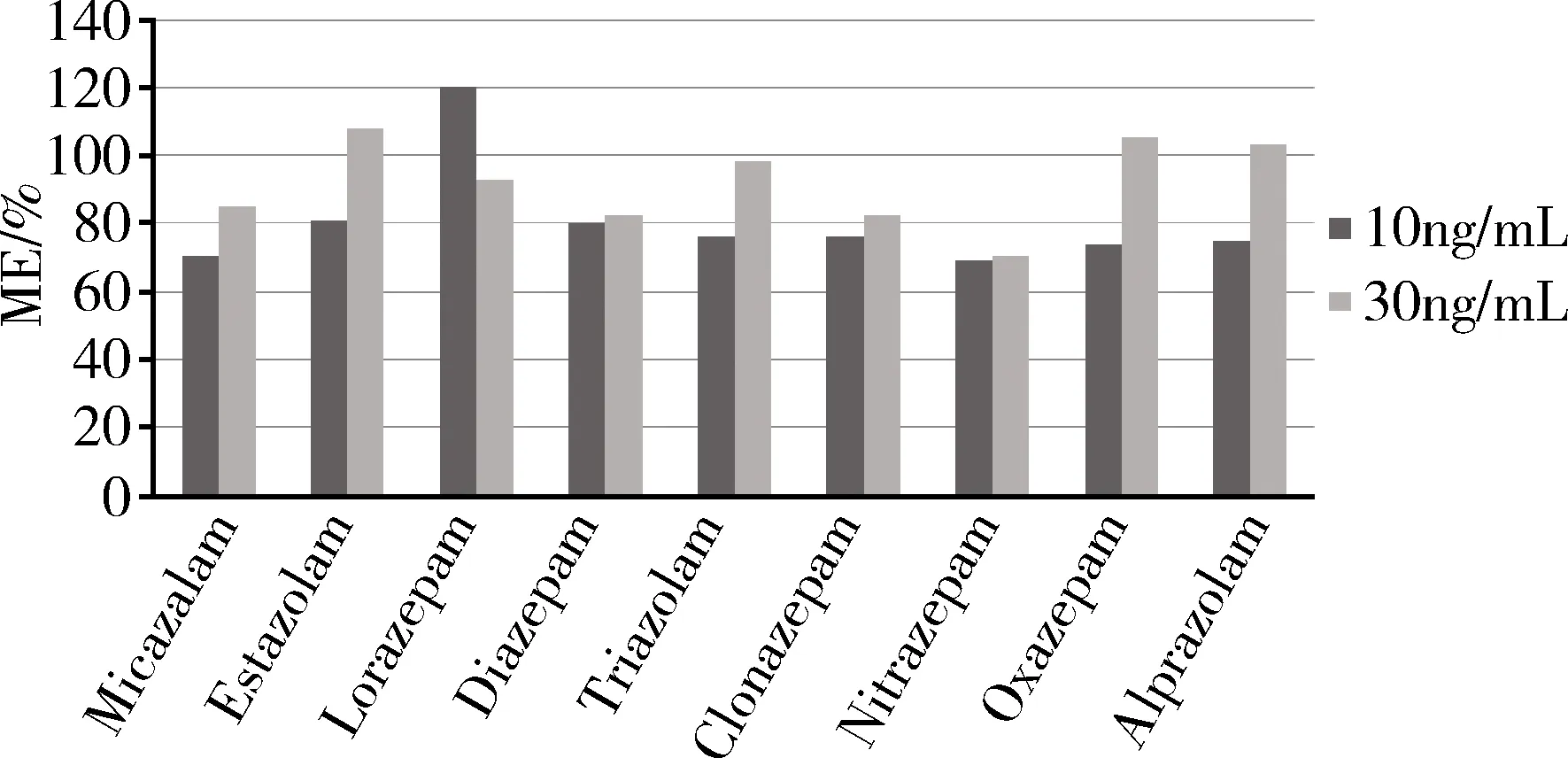
图2 9种苯二类药物的基质效应Fig 2 Matrix effect of nine benzodiazepine drugs
表2 9种苯二类药物的线性关系、检出限和定量限Tab 2 Linear relationship, LOD and LOQ of nine benzodiazepine drugs
2.5 回收率及相对标准偏差考察 取空白样品,分别按10、50、100 μg/kg 添加3个水平的混合对照品溶液,考察方法的回收率和日间、日内相对标准偏差(RSD)。每个添加水平制备平行样6份,在1.3.3项和1.4项条件下进行前处理和上机分析,计算相应的回收率与相对标准偏差(Relative Standard Deviation,RSD),结果显示,在3个加标水平下,9种目标分析物的加标平均回收率为69.1%~108.0%,日内RSD为1.8%~9.0%,日间RSD为2.6%~9.3%(表3)。
表3 9种苯二类药物的平均回收率与相对标准偏差(n=6)Tab 3 Average recovery and RSD of nine benzodiazepine drugs(n=6)
3 讨论与结论