Microglia depletion as a therapeutic strategy: friend or foe in multiple sclerosis models?
2022-06-29VictoriaSofiaBereniceWiesManciniAnabellaAyelenDiPietroLauraAndreaPasquini
Victoria Sofia Berenice Wies Mancini , Anabella Ayelen Di Pietro , Laura Andrea Pasquini
Abstract Multiple sclerosis is a chronic central nervous system demyelinating disease whose onset and progression are driven by a combination of immune dysregulation, genetic predisposition, and environmental factors. The activation of microglia and astrocytes is a key player in multiple sclerosis immunopathology, playing specific roles associated with anatomical location and phase of the disease and controlling demyelination and neurodegeneration. Even though reactive microglia can damage tissue and heighten deleterious effects and neurodegeneration, activated microglia also perform neuroprotective functions such as debris phagocytosis and growth factor secretion. Astrocytes can be activated into pro-inflammatory phenotype A1 through a mechanism mediated by activated neuroinflammatory microglia, which could also mediate neurodegeneration. This A1 phenotype inhibits oligodendrocyte proliferation and differentiation and is toxic to both oligodendrocytes and neurons. However, astroglial activation into phenotype A2 may also take place in response to neurodegeneration and as a protective mechanism. A variety of animal models mimicking specific multiple sclerosis features and the associated pathophysiological processes have helped establish the cascades of events that lead to the initiation, progression, and resolution of the disease. The colonystimulating factor-1 receptor is expressed by myeloid lineage cells such as peripheral monocytes and macrophages and central nervous system microglia. Importantly, as microglia development and survival critically rely on colony-stimulating factor-1 receptor signaling, colony-stimulating factor-1 receptor inhibition can almost completely eliminate microglia from the brain. In this context, the present review discusses the impact of microglial depletion through colony-stimulating factor-1 receptor inhibition on demyelination, neurodegeneration, astroglial activation, and behavior in different multiple sclerosis models, highlighting the diversity of microglial effects on the progression of demyelinating diseases and the strengths and weaknesses of microglial modulation in therapy design.
Key Words: astrocytes; colony-stimulating factor-1 receptor inhibition; cuprizone; demyelination; microglia; multiple sclerosis; neurodegeneration

Introduction 267 Search Strategy and Selection Criteria 267 Microglial Activation in Demyelinating Diseases 268 Microglial Depletion as an Experimental Strategy 268 Final Considerations 270
Introduction
Multiple sclerosis (MS) is a chronic central nervous system (CNS) demyelinating disease whose onset and clinical progression are driven by a combination of genetic factors, dysregulated immunity, and environmental cues (Thompson et al., 2018). Late-stage clinical symptoms are a consequence of early axonal and neuronal damage induced by an association of inflammatory mediators, demyelination, and loss of trophic support, in combination with an inflammatory process confined to the CNS and driven by astrocytes and microglia (Correale et al., 2017). An estimated 85% of patients are affected by relapsing-remitting MS, a disease presentation characterized by recurrent symptoms and subsequent total or partial recovery. Later on, up to 50% of untreated relapsing-remitting MS patients may present secondary progressive MS, which involves relentless clinical deterioration. The remaining 15% of patients, however, show primary progressive MS, consisting of progressive deterioration from disease onset. Current treatments for relapsing-remitting MS and secondary progressive MS target immune system suppression to ameliorate the severity and reduce the frequency of new relapses. In contrast, therapeutic options for primary progressive MS are still scarce and remain to be addressed (Faissner et al., 2019). Given that immune-suppressing drugs are only effective in relapsing-remitting MS but not in progressive MS, it is plausible that differential pathogenic mechanisms involved in these MS forms may guide the development of alternative therapeutic agents. Supporting this view, evidence shows that the immune mechanisms of progressive MS are predominantly driven by resident CNS cells, whereas those present in MS relapses are triggered by transient infiltration of peripheral immune cells (Healy et al., 2022).
A wide range of animal models mimicking the specific features and pathophysiological processes of MS have helped establish the cascades of events that could lead to disease onset, clinical course, and resolution. Experimental demyelination models are mediated by immunity, viruses, and toxins. For instance, experimental autoimmune encephalomyelitis (EAE), the most widely used animal model of CNS demyelination, is induced by immunization with myelin proteins and is particularly useful to study the autoimmunity features of MS, while also reproducing some MS motor disabilities. Unfortunately, although numerous therapeutic strategies demonstrated beneficial effects in the EAE model, they have yielded poor or no beneficial results in MS pathology. In contrast, toxin models are especially useful in the evaluation of therapeutic agent effects on demyelination and remyelination processes. These models comprise (1) focal demyelination using, for example, lysolecithin or ethidium bromide, and (2) systemic toxin administration. Among the latter, cuprizone (CPZ) administration is an increasingly used demyelination model, which lacks the T cell infiltration of autoimmune response-mediated models. CPZ demyelination is characterized by mature oligodendrocyte loss and demyelination concomitant with microglial and astroglial activation (Figure 1A1
andB1
). This model has been used in two protocols: (i) an acute model of 5 or 6 weeks of CPZ administration to adult mice, which produces demyelination followed by spontaneous remyelination upon CPZ withdrawal, and (ii) a chronic model of 12-week CPZ feeding, which fails to induce remyelination after CPZ withdrawal and thus leads to neurodegeneration (Figure 1B1
). Given that, as mentioned above, the mechanisms underlying progressive MS involve resident CNS cells –but not peripheral immune cells (Healy et al., 2022)– and lead to neurodegeneration, prolonged CPZ administration could be thought to mimic progressive forms of MS (Wies Mancini et al., 2019). It is worth highlighting that, despite the advantages and disadvantages of widely established animal models, they fail to single-handedly replicate MS stages and should actually be regarded as complementary.Search Strategy and Selection Criteria
The literature cited in this review was published between 2006 and 2022 and searched on the PubMed database (www.pubmed.ncbi.nlm.nih.gov) using a combination of the following terms: depletion, activation, microglia, astrocytes, neurons, oligodendrocytes, multiple sclerosis, experimental models, cuprizone, demyelination, remyelination, neurodegeneration, inflammation, behavior, CSF1-R inhibition, and BLZ945. Search results were further filtered by title, abstract, and year.
Microglial Activation in Demyelinating Diseases
Microglia play a key role in the maintenance of CNS homeostasis. Microglia communicate with neurons and non-neural cells to regulate some of their functions, thus contributing to the healthy development of the neural network and synaptogenesis through the release of neurotrophic factors. In addition, microglia participate in neuronal repair and brain circuit remodeling through their phagocytic capacity. In terms of their well-known immune function, microglia constantly survey their microenvironment and are activated in response to injury or disease, undergoing profound morphological and transcriptional changes to promote repair (Han et al., 2019; Tay et al., 2017;Figure 1C
). In the normal inflammatory response, microglia return to a surveillance state after activation (Writght-Jin et al., 2019). In a pathological scenario, phagocytic microglia scavenge the CNS for cellular debris and pathogens, giving way to repair processes (Galloway et al., 2019). In particular, during demyelination, microglia participate in myelin debris clearance through phagocytosis, a crucial step for oligodendroglial differentiation and remyelination (Kotter et al., 2006; Wies Mancini et al., 2019;Figure
1B1
). However, in the context of chronic and severe damage in neurological diseases such as MS, microglia fail to return to a surveillance state and remain continuously activated. Sustained and exacerbated microglial activation has negative effects on the CNS which lead to neurodegeneration, thus worsening long-term clinical neurological symptoms in MS. Specifically, although activated microglia attempt to contain the damage by producing antiinflammatory factors, they also secrete toxic pro-inflammatory substances and induce astrocytes reactivity. These events impair remyelination and promote the release of reactive oxygen/nitrogen species which cause neuronal oxidative damage and increase neuroinflammation. In addition, this activation is maintained by the adenosine triphosphate produced by neuronal death and by the inhibition of transforming growth factor beta signaling caused by activated microglia themselves (Faissner et al., 2019; Han et al., 2019). In homeostasis, axons with thinner myelin sheaths have larger axonal mitochondria, which reflects their higher metabolic status. This link between myelin sheath thickness and mitochondrial size is temporarily lost along with demyelination but later reestablished in advanced remyelination (Ineichen et al., 2020). Demyelination increases axonal energy demand and interferes with axonal transport, especially in mitochondria, which induces axonal dysfunction and alterations in metabolism and ion channels (Nasi et al., 2020;Figure
1B1
). Toll-like receptor-activated microglia can promote different degrees of neuronal network dysfunction, with severe dysfunction being caused mostly by reactive oxygen/nitrogen species rather than proinflammatory cytokines. Supporting evidence has shown the prevention of neuronal disturbance through either microglial depletion or the pharmacological inhibition of oxidant-producing enzymes, inducible nitric oxide synthase, and nicotinamide adenine dinucleotide phosphate oxidase (Schilling et al., 2021).Microglia-astrocyte communication in demyelination and
neurodegeneration
Astrocytes are also key to immune surveillance, the maintenance of neurotransmitter pools, metabolism, trophic support, myelin sheath formation, synaptic formation and plasticity, and injury healing (Manninen et al., 2020). Astrocytes take up glutamate at synapses in response to neuronal activity, which induces aerobic glycolysis and the secretion of lactate to be consumed by neurons (Beard et al., 2022). In particular, astrocytes express glutamate transporters to sense variations in neuronal activity at the synapse level and express glucose transporter 1 at the vasculature level to allow glucose uptake (Patching, 2017). Astrocytes further facilitate glucose uptake by releasing vasoactive substrates in response to neuronal activity (Beard et al., 2022). Notably, neurons and astrocytes show specificity in the predominance of their metabolic pathways, with a glycolytic profile in astrocytes and an oxidative profile in neurons (Figure 1D
). Neurodegenerative diseases associated with aging evidence diminished brain energy consumption in specific regions, which leads to cognitive impairment, and alterations in neuronal function and excitability (Muddapu et al., 2020). Supporting data obtained from both animal and human studies have shown that aging is accompanied by a decrease in aerobic glycolysis in astrocytes and mitochondrial oxidative phosphorylation in neurons (Goyal et al., 2017). Moreover, aging has been linked to the loss of glucose transporters, which leads to synaptic dysfunction and susceptibility to neuronal degeneration (Beard et al., 2022;Figure 1E
).As in other disorders, astrocytes become reactive after CNS demyelination, which entails astroglial hypertrophy and intermediate filament protein upregulation such as glial fibrillary acidic protein and vimentin. CPZ-induced demyelination and remyelination studies have shown the need for microglia in the appearance of reactive astrocytic phenotypes (Marzan et al., 2021). Microglia-derived interleukin-1β (IL-1β) can regulate ciliary neurotrophic factor and induce astroglial activation, which promotes fiber myelinationin vitro
. IL-1β also stimulates astroglial production of leukemia inhibitory factor, thus promoting oligodendrocyte survival and ameliorating EAE (Traiffort et al., 2020). Abundant evidence supports the notion that reactive astrocyte actions are involved in spontaneous remyelination, which may be remarkably efficient both in experimental demyelination models and MS patients (Franklin and Ffrench-Constant, 2017). However, remyelination usually fails or takes place in the plaque periphery in chronic lesions, where oligodendroglial progenitor migration and/or differentiation are probably affected (Franklin and Ffrench-Constant, 2017). This unsuccessful remyelination has also been associated with deleterious astroglial activity, mostly in terms of more severe reactivity and the secretion of harmful molecules. Among these molecules, tumor necrosis factor α has been mainly identified in fibrous astrocytes in the peripheral region of chronic active MS lesions. In addition, the interferon γ (IFN-γ) receptor expressed in astrocytes has been implicated in the expression of chemokines and infiltration by inflammatory cells, which induce demyelination in both Th1- and Th17-mediated adoptive EAE (Loos et al., 2020). Moreover, ectopic astroglial expression of IFN-γ delays recovery in EAE mice, while CNS delivery of IFN-γ produces a sharp reduction in oligodendroglial repopulation in the CPZ model. Concomitantly, IFN-γ seems to play a key role in MS lesion progression. Among molecules highly expressed by reactive astrocytes, endothelin-1 is considered a potent inhibitor of remyelination (Traiffort et al., 2020).In addition to growth factors, cytokines, and neuropeptides, reactive astrocytes also secrete extra-cellular matrix molecules which significantly modify the MS lesion environment and affect oligodendroglial precursor behavior (Pu et al., 2018). Both the receptors present on the surface of oligodendroglial precursors and the astrocyte-secreted extra-cellular matrix molecules can shift the environment from remyelination-permissive to -inhibitory. As a matter of fact, except for the interaction between astroglial laminin and oligodendroglial α6β1 integrin, which attenuates oligodendrocyte deathin vitro
(Traiffort et al., 2020). Extra-cellular matrix accumulation appears to be a key factor explaining the failure of tissue regeneration.Astroglial activation could certainly mediate neurodegeneration; in certain scenarios, astrocytes are activated into pro-inflammatory phenotype A1, which is induced by classically activated neuroinflammatory microglia, inhibits oligodendroglial precursor proliferation and differentiation, and is toxic to neurons and oligodendrocytes (Liddelow et al., 2017). However, astrocytes may be activated into phenotype A2 as a result of neurodegeneration and as a protective mechanism. In MS lesions, A1 astrocytes have been mainly detected in the active lesions, whereas A2 astrocytes have been found along remyelination (Haindl et al., 2019). Furthermore, the depletion of astrocytes through intracallosal injection of L-a-aminoadipate in CPZ mice promotes a significant expansion of the myelinated areas, a reduction in Iba-1microglial staining, and a sharp decrease in the expression of genes associated with either microglia recruitment, typically triggered by astrocytes, or the loss of oligodendroglial precursor differentiation (Madadi et al., 2019).
Microglial heterogeneity in response to demyelination and neurodegeneration
New tools designed to distinguish microglia from other myeloid populations have unveiled the diversity of microglial functions and phenotypes (Masuda et al., 2019). Microglia possess region-specific transcriptional profiles and differential expression of immunoregulatory proteins which highlight their heterogeneity. This region-specific diversity allows microglia to fulfill specialized homeostatic functions and a variety of responses in different CNS pathologies (Plastini et al., 2020).
Microglial activation is linked to active demyelination and neurodegeneration in MS and has therefore been associated with early axonal damage and disease progression (Zrzavy et al., 2017). Microglia are grouped in clusters or nodules in early lesion formation when demyelination is not yet observed (normal-appearing white matter). These pre-active or early active lesions containing activated microglia are not accompanied by blood-brain barrier alterations or astrogliosis but have shown an association with degenerating axons. Later, active white matter (WM) lesions show an increase in microglial activation, the loss of their homeostatic signature, and close contact of microglial processes with transected axons (Zrzavy et al., 2017). Chronic active WM lesions contain a hypocellular demyelinated center surrounded by CD68microglia/macrophages with residual lipids. In contrast, chronic inactive demyelinated WM lesions are hypocellular but rarely exhibit residual CD68microglia/macrophages (Kuhlmann et al., 2017). In grey matter (GM) lesions, microglial activation has been linked with cortical demyelination and neurodegeneration (Jafari et al., 2021). In this case, microglia show a gradient pattern with higher activation in layers of the meningeal surface vicinity, where GM damage is more substantial, and lower activation in the deeper cortex (Magliozzi et al., 2013).
Single-nucleus transcriptomic studies of post-mortem MS tissue have also evidenced the functional diversity of microglia, the loss of their homeostatic signature, and a wide range of activation phenotypes that dynamically change with disease evolution (Schirmer et al., 2019). In particular, microglia upregulate glycolysis and iron homeostasis gene expression in GM but increase lipid metabolism gene expression in WM. In other words, microglia show differences in gene signatures between GM and WM and region-specific functions (van der Poel et al., 2019).
Microglial Depletion as an Experimental Strategy
Microglia depletion has been evaluated in different neurodegenerative diseases as a strategy to reduce neuroinflammation and design novel therapies. Microglia may be depleted by means of genetic strategies, for instance, by activating the suicide gene HSVTK under the CD11b promoter (Han et al., 2019). Given that chemokine receptor CX3CR1 is primarily expressed in microglia in the CNS, genetic modulation of microglia using CX3CR1-Cre lines is also a common depletion method. In this case, depletion may involve inducibly expressed diphtheria toxin A (DTA) in CX3CR1CreER/+: R26iDTA/+, or iDTA mice, or inducibly knocked out colony-stimulating factor-1 receptor (CSF-1R) in CX3CR1CreER/+:Csf1rFlox/Flox mice (Willis and Vukovic, 2020; Wu et al., 2020), However, the most widely used strategy is the pharmacological inhibition of CSF-1R, a class III tyrosine kinase receptor which has two ligands, CSF-1 and IL-34, and is expressed by myeloid lineage cells (Chitu et al., 2016). Given their great dependence on CSF-1R signaling for development and survival, microglia can be mostly depleted from the brain by means of CSF-1R inhibitors (Figure 1A2
andB2
).Pharmacological CSF-1R inhibition to deplete microglia: drugs and
selectivity
Reliable conclusions on the use of CSF-1R inhibitors require a complete characterization of inhibitor effects on other CNS cell types. Liu et al. (2019) have addressed this point, showing that CSF-1R inhibitors have different degrees of specificity and can also bind to other tyrosine kinase receptors like platelet-derived growth factor receptor α, highly expressed by oligodendroglial progenitors and essential for their survival. PLX5622 and PLX3397 are two potent CSF-1R inhibitors with IC50 of 10 and 20 nM, respectively. High doses of both PLXs deplete microglia but also affect the number of oligodendroglial progenitors, while only low doses of PLX5622 selectively affect microglia. In turn, 4-[2((1R,2R)-2-hydroxycyclohexylamino)-benzothiazol-6-yloxyl]-pyridine-2- carboxylic acid methylamide (BLZ945) has been widely used as a therapeutic agent in disorders associated to the monocytic lineage (Webb et al., 2018; Lu et al., 2019; Liaw et al., 2020; Crotty et al., 2021; Fang et al., 2021; Pfirschke et al., 2022). As different from PLXs, BLZ945 does not affect the viability of oligodendroglial progenitorsin vitro
, which may be due to its higher specificity, i.e., an IC50 under 1 nM for CSF-1R and over 1 µM for platelet-derived growth factor receptor β. These data may explain the contrasting results obtained using different CSF-1R inhibitors, which may be a consequence of administration methods, formulation, dosage, duration of treatment, and pharmacokinetics affecting cells other than microglia (Han et al., 2019).Beneficial effects of pharmacological microglial removal
Regarding the beneficial effects of microglial depletion, experimental models of Alzheimer’s disease have shown a reduction in amyloid deposits, Aβ plaque formation (Sosna et al., 2018) and neuronal loss, and an improvement in memory functions (Spangenberg et al., 2019; Gratuze et al., 2021). Microglial depletion has further been shown to improve neuropathic pain through lower pro-inflammatory cytokine expression (Lee et al., 2018; Wang et al., 2018). In models of demyelinating diseases, therapeutic or prophylactic CSF-1R inhibition has been found to curb disease severity and enable a more permissive environment for remyelination and recovery in a mouse model of EAE (Lassmann and Bradl, 2017; Nissen et al., 2018). Likewise, CSF-1R inhibitor GW2580 slowed the progression of EAE and decreased clinical scores in a rat model (Borjini et al., 2016), while CSF-1R stimulation with its ligands CSF-1 or IL-34 increased protective CD11cmicroglia and attenuated EAE symptoms (Wlodarczyk et al., 2015). In addition, using PLX3397 in mice carrying mutations in the oligodendrocyte PLP1 gene as an MS model, Groh et al. (2019) demonstrated that microglial depletion reduces neuroinflammation and CNS T cell recruitment, which ameliorates demyelination, axonal damage, and neuronal loss. Tahmasebi et al. (2019) used PLX3397 treatment in a chronic CPZ-induced demyelination model and observed improved remyelination and a reduction in fiber damage and inter-sheath area, concomitant with motor recovery. Moreover, Marzan et al. (2021) applied PLX3397 treatment in acute CPZ-induced demyelination and found microglial depletion to prevent demyelination, oligodendroglial loss, and reactive astrocytosis. Ultrastructural studies by electron microscopy initially indicated that myelin sheaths remained intact in CPZ-treated mice upon microglial depletion. However, these sheaths were vigorously phagocytosed upon microglial repopulation, which revealed CPZ-induced myelin damage. These results appeared to initially correlate with those obtained by Lampron et al. (2015) in CX3CR1mice showing resistance to CPZ-induced demyelination, although this observation actually resulted from unsuccessful myelin debris phagocytosis leading to aberrant remyelination and the loss of axon integrity.
Detrimental effects of pharmacological microglial removal
On the other hand, evidence has also been documented of the detrimental effects of microglial depletion. It has been reported that microglial clearance does not affect the number of amyloid plaques in models of Alzheimer’s disease but leads to increased plaque size (Zhao et al., 2017). Furthermore, microglial clearance has also exacerbated cerebral neurotoxicity in cerebral ischemia, Parkinson’s disease, and coronavirus encephalitis (Janda et al., 2018; Wheeler et al., 2018; Yang et al., 2018; Wen et al., 2020; Jia et al., 2021). More recently, Tanabe et al. (2019) used EAE in non-obese diabetic mice as a model of secondary progressive MS and showed that microglial depletion by PLX3397 significantly worsened secondary disease progression, increased mortality rates, promoted inflammation, and spurred CD4T cell proliferation, which led to exacerbated demyelination and axonal degeneration. Finally, microglial depletion through PLX5622 treatment has been shown to exacerbate demyelination and impair remyelination in neurotropic coronavirus infection (Sariol et al., 2020).
Consequences of microglial depletion on neuron-microglia crosstalk
Among other functions, microglia play a fundamental role in the support of neuronal functions. In physiological conditions, microglia and neurons maintain bidirectional communication from embryonic development to adulthood. The CX3CL1-CX3CR1 and CD200-CD200R axes are key in maintaining microglia in a resting state through permanent crosstalk with neurons and modulating microglia behavior in both physiological and pathological scenarios (Pawelec et al., 2020; Manich et al., 2019). In both CX3CL1-CX3CR1 and CD200-CD200R, receptors are present in microglia while the ligand is secreted by neurons. CSF-1R inhibition interrupts this dialog and thus blocks microglial protection of neuronal function, which could lead to neurodegeneration.
Analogously, in physiological conditions, neuronal CSF-1 prevents inappropriate microglia activation and neurotoxicity. High levels of CSF-1 and microgliosis have been observed in numerous CNS disorders promoting either neuronal survival or tissue damage (Chitu et al., 2016). Direct injection of CSF-1 into WM has been shown to produce focal microgliosis and demyelination (Marzan et al., 2021), and high levels of CSF-1R and ligand CSF-1 have been detected in CNS tissue from MS patients. Furthermore, CSF-1R may be regarded as a key node of MS disease progression; indeed, using a potent small-molecule CSF-1R inhibitor to block phosphorylation and downstream signaling, Hagan et al. (2020) succeeded in ameliorating neuroinflammation and curbing microglia proliferation in a murine lipopolysaccharide model, and in preventing axonal and neurological damage in EAE. These studies support the potentially beneficial effect of downstream CSF-1R signaling modulation in the context of CNS injury.
Our group has used orally administered brain-penetrating inhibitor BLZ945 in the CPZ model to evaluate thein vivo
impact of microglia depletion on demyelination, remyelination, and neurodegeneration, as well as its association with astroglial activation and behavioral changes. Results showed that preventive BLZ945 administration ameliorated demyelination in the acute CPZ protocol, mostly in the cortex and external capsule, but failed to preserve myelin or promote remyelination in myelin-rich areas, which reflects the loss of microglia phagocytic capacity and its negative consequences in oligodendroglial differentiation. Preventive and therapeutic BLZ945 treatment protected myelin and favored remyelination in the chronic CPZ protocol (Wies Mancini et al., 2019;Figure 1B2
). However, in this case, BLZ945 treatment aggravated neurodegeneration, as evidenced by several terminal axonal ovoids but no neuronal body loss (Figure 1B2
). In addition, neurodegeneration closely correlated with increased astroglial activation (Wies Mancini et al., 2022), which may be considered an attempt to induce microglia migration and recruitment interrupted by BLZ945 (Figure 1B2
). Previous evidence has demonstrated that astrocytes can release extracellular signals, particularly chemokine CXCL10, and recruit microglia to remove myelin debris in demyelination processes. CXCL10 is detectable around active MS lesions; moreover, CXCL10 production in EAE increases before the appearance of symptoms and remains high at the most severe stages of the disease, later decreasing along with remission (Traiffort et al., 2020). This evidence is in keeping with our findings showing an increase in CXCL10 mRNA expression in CPZ-demyelinated, BLZ945-treated animals.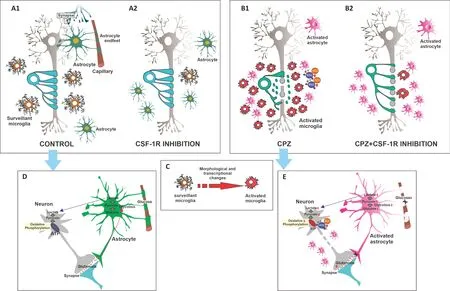
Figure 1|Consequences of CSF-1R-induced-microglial depletion on cuprizone-induced demyelination in central nervous system.Administration of cuprizone (CPZ) is characterized by severe loss of mature oligodendrocyte and demyelination, accompanied by microglia and astrocyte activation and axonal degeneration (A1 vs. B1). Microglia respond to most neurological disorders through activation mediated by profound morphological and transcriptional changes to perform neuroprotective functions, such as debris phagocytosis and growth factor secretion (C). However, sustained and exacerbated microglia activation can damage tissue and contribute to neurodegeneration (B1). Microglial activation in MS has been linked to early axonal damage and the progression of the disease. These persistent dysregulated microglia can induce a pro-inflammatory astrocyte phenotype called A1, which inhibits oligodendroglial precursor proliferation and differentiation and has toxic effects on neurons and oligodendrocytes (B1). These effects are mediated by an increase in reactive oxygen/nitrogen species (ROS/RNS), which leads to mitochondrial damage (D vs. E). Astrocytes maintain neurotransmitter pools, provide trophic support, and take part in metabolism, synaptic formation and plasticity, myelin sheath formation, injury healing, and immune surveillance (D). Astrocytes take up glutamate at synapses in response to neuronal activity, which triggers aerobic glycolysis and lactate secretion to be consumed by neurons (D). Astrocytes express glutamate transporters to sense changes in neuronal activity at the synapse level and glucose transporter 1 (GLUT1) at the vasculature level to allow glucose uptake (D). Moreover, astrocytes further facilitate glucose uptake by releasing vasoactive substrates in response to neuronal activity (D). Neurodegeneration is associated with decreased aerobic glycolysis in astrocytes, mitochondrial oxidative phosphorylation in neurons, and the loss of glucose transporters, which leads to synaptic dysfunction and susceptibility to neuronal degeneration (E). Demyelination increases axonal energy demand and causes poor axonal transport, especially in mitochondria, which leads to axonal dysfunction with metabolic alterations and ion channel disturbances (E). Microglia can be almost completely depleted from the brain using colony-stimulating factor-1 receptor (CSF-1R) inhibitors such as BLZ945 (BLZ) (A2). CSF-1R inhibition through BLZ treatment successfully reduced the microglial population and myelin loss in the chronic CPZ model, but also exacerbated axonal degeneration (B2). This axonal degeneration was accompanied by mild behavioral alterations but no neuronal body loss (B2). These results should be taken into account when proposing the modulation of microglial activation in the design of therapies relevant for demyelinating diseases.
Behavioral alterations in MS and the CPZ model: effects induced by
microglial depletion
Motor and cognitive alterations are present in 80% of MS patients and include diminished balance and walking speed, loss of hand and foot dexterity, slower information processing, and poorer episodic memory as the most prevalent (Pellegrino et al., 2018). Evidence has shown that motor deficits are linked to CNS demyelination, mostly in the cerebellum and spinal cord (D’Ambrosio et al., 2017; Wilkins, 2017; Parmar et al., 2018). As pain and fatigue, common symptoms of MS, are not observed in the CPZ model, motor alterations are frequently used in this model to evaluate functional involvement, although with sparse results (Sen et al., 2019). While increasing test complexity through ladder crossing or wheel running seems to be a promising strategy to detect early subtle deficiencies, dissecting motor deficits from cognitive ones may prove a challenge. Moreover, CPZ-fed animals exhibit high-stress hormone and neurotransmitter levels, which makes them more active and may thus lead to behavioral misevaluation.
Our studies (Wies Mancini et al., 2022), using mostly the same tests as in previous reports (Xiu et al., 2017; Chang et al., 2017), barely showed behavioral alterations resulting from axonal degeneration in CPZ or CPZ+BLZ945 animals. Indeed, unaltered behavior is in line with the NeuNcell results, which rendered no differences across experimental groups, and with amino-cupric-silver staining results, which only showed axonal tract degeneration and almost no apoptotic neuronal bodies. Y-maze test results indicate that chronic CPZ administration does not affect locomotion or working memory, as arm entries can be regarded as an animal mobility indicator. However, at the end of both the acute and the chronic protocols, CPZ animals outperformed CPZ+BLZ945 ones (Wies Mancini et al., 2022). These results are in agreement with findings reported by Torres et al. (2016), who showed poorer performance in animals fed 290 mg/kg PLX3397 for 21 days than in controls. Moreover, the authors detected no significant differences between experimental conditions in the open field test, which indicates comparable locomotor activity.
In addition, our results on recognition memory show longer exploration of new objects than familiar ones in all experimental conditions. Although CPZ animals devoted significantly less time to exploring the new object than controls, a recovery toward control values was recorded in the CPZ + BLZ945 group (Wies Mancini et al., 2022). The administration of CSF-1R inhibitors at high doses has been shown to eliminate microglia and affect spatial memory in murine models, with unaltered social behavior (Torres et al., 2016). Microglia-derived brain-derived neurotrophic factor plays a major role in memory consolidation, and alterations in neuron-microglia communication appear to disrupt behavior (Torres et al., 2016). Altogether, these results may be thought to unveil compensatory mechanisms which prevent at least observable behavioral alterations.
Acute MS lesions entail mild permanent disability, as the CNS compensatory capacity helps most axons overcome acute demyelination. However, with disease progression, an additional neuronal loss can no longer be compensated for, which gives way to progressive MS. For these reasons, sectioned axons and axonal ovoids are more frequently observed in chronic than in acute lesions (Mahad et al., 2015). In this regard, the mild behavioral alterations associated with BLZ945- and CPZ-induced axonal degeneration could be associated with both acute MS lesions and the onset of chronic ones. However, longer treatments may be necessary to study the loss of CNS compensatory capacity against demyelination.
Caveats of MG depletion
It should be pointed out that, despite the large number of studies evaluating microglia depletion as a treatment for neurological diseases, many questions remain unanswered, especially considering differential microglial activation in the different types of disease processes. In addition, studies, where microglial depletion was applied in preventive protocols, do not seem to be as useful as those where treatment began after the model disease was induced. Moreover, current research shows that CSF-1R kinase inhibitors may target several cell populations other than microglia, including meningeal, perivascular, and choroid plexus macrophages and microglial progenitor cells (Yang et al., 2018). Therefore, the notion of pure microglial depletion using the systemic delivery of currently available CSF-1R inhibitors should be thoroughly examined and cautiously interpreted. Most importantly, the possible side effects of microglial depletion should be considered. In nonsterile conditions, depletion may trigger at least transient immunodeficiency, which might expose the CNS to infection and disrupt CNS homeostatic function in unaffected brain areas. Microglial depletion followed by microglial repopulation thus seems to be a more plausible therapeutic approach, while the replacement of dysfunctional or aberrantly activated microglia in the areas affected may constitute the most efficient strategy.
Final Considerations
Using mRNA sequencing,in situ
hybridization of individual cells, and immunohistochemistry, Masuda et al. (2019) have recently characterized microglia subtypes in several areas of the CNS in development and disease. The authors have shown different disease-related subclasses of microglia which coexist in the brain of MS patients and whose gene expression profiles are phenotypically similar to mouse microglia subclasses in CPZinduced demyelination. Our results further show differential effects of CPZ demyelination and microglial depletion through BLZ945 on myelin protection and axonal degeneration across different CNS areas, which may reflect microglial region-dependent heterogeneity (Wies Mancini et al., 2022). Worth pointing out, astrocytes (Liddelow et al., 2017) and oligodendrocytes (Foerster et al., 2019) have also shown high regional heterogeneity, which may also explain the different responses observed across CNS regions along with demyelination and neurodegeneration.The worsening of myelin debris phagocytosis upon microglial depletion reflects the central role of microglia in demyelination. Insufficient phagocytosis subsequently hinders remyelination, particularly in myelinrich areas, and leads to neurodegeneration. Astrocytes also take part in myelin uptake, especially as an early response to damage which ultimately triggers immune cell recruitment (Ponath et al., 2017). This early response may have positive or negative effects on lesion pathology depending on the inflammatory environment, which is itself altered by microglia depletion. Strikingly, oligodendroglial populations which express genes involved in antigen processing and presentation (major histocompatibility complex-I and -II) have been recently characterized in the EAE model. Furthermore, oligodendroglial precursors have been recently reported to have the phagocytic capacity, while those expressing major histocompatibility complex-II have been shown to activate memory and effector CD4T cells (Falcão et al., 2018). Overall, microglial depletion may disrupt the inflammatory scenario of demyelinating lesions, promoting beneficial or harmful responses by astrocytes and oligodendrocytes which impact neurodegeneration.
In summary, the present review focuses on the role of microglia and astrocytes in demyelination, remyelination, and neurodegeneration, and discusses the consequences of microglial depletion through CSF-1R inhibition on demyelination, neurodegeneration, astrocyte activation and behavior in different MS models. The findings reported here highlight the diversity of microglial effects on the progression of demyelinating diseases and the importance of ongoing research for the development of appropriate therapies in neurodegenerative pathologies such as MS.
Author contributions:
VSBWM and AADP collected evidence and wrote the manuscript draft. LAP wrote the final version of the manuscript and designed the figure. All authors approved the final version of the manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Karl E. Carlström, Karolinska Institutet, Sweden.
Additional file:
Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- c-Abl kinase at the crossroads of healthy synaptic remodeling and synaptic dysfunction in neurodegenerative diseases
- The mechanism and relevant mediators associated with neuronal apoptosis and potential therapeutic targets in subarachnoid hemorrhage
- Brain and spinal cord trauma: what we know about the therapeutic potential of insulin growth factor 1 gene therapy
- Functions and mechanisms of cytosolic phospholipase A2 in central nervous system trauma
- Cre-recombinase systems for induction of neuronspecific knockout models: a guide for biomedical researchers
- Prenatal programing of motivated behaviors: can innate immunity prime behavior?