The mechanism and relevant mediators associated with neuronal apoptosis and potential therapeutic targets in subarachnoid hemorrhage
2022-06-29QiTianShengLiuShouMengHanWeiZhangXianYaoQinJunHuiChenChengLiLiuYuJiaGuoMingChangLi
Qi Tian, Sheng Liu, Shou-Meng Han, Wei Zhang, Xian-Yao Qin, Jun-Hui Chen, Cheng-Li Liu, Yu-Jia Guo, Ming-Chang Li
Abstract Subarachnoid hemorrhage (SAH) is a dominant cause of death and disability worldwide. A sharp increase in intracranial pressure after SAH leads to a reduction in cerebral perfusion and insufficient blood supply for neurons, which subsequently promotes a series of pathophysiological responses leading to neuronal death. Many previous experimental studies have reported that excitotoxicity, mitochondrial death pathways, the release of free radicals, protein misfolding, apoptosis, necrosis, autophagy, and inflammation are involved solely or in combination in this disorder. Among them, irreversible neuronal apoptosis plays a key role in both short- and long-term prognoses after SAH. Neuronal apoptosis occurs through multiple pathways including extrinsic, mitochondrial, endoplasmic reticulum, p53 and oxidative stress. Meanwhile, a large number of blood contents enter the subarachnoid space after SAH, and the secondary metabolites, including oxygenated hemoglobin and heme, further aggravate the destruction of the blood-brain barrier and vasogenic and cytotoxic brain edema, causing early brain injury and delayed cerebral ischemia, and ultimately increasing neuronal apoptosis. Even there is no clear and effective therapeutic strategy for SAH thus far, but by understanding apoptosis, we might excavate new ideas and approaches, as targeting the upstream and downstream molecules of apoptosis-related pathways shows promise in the treatment of SAH. In this review, we summarize the existing evidence on molecules and related drugs or molecules involved in the apoptotic pathway after SAH, which provides a possible target or new strategy for the treatment of SAH.
Key Words: blood-brain barrier; mechanism; mediators; neuronal apoptosis; pathways; subarachnoid hemorrhage; targets; treatment

Introduction 244 Retrieval Strategy 244 Apoptosis Pathways 244 Apoptosis in Early Brain Injury 246 Apoptosis in Delayed Cerebral Ischemia and Cerebral Vasospasm 246 Blood-Brain Barrier Dysfunction and Cerebral Edema Facilitate Apoptosis 247 Treatment 247 Conclusion 248
Introduction
Subarachnoid hemorrhage (SAH) is a type of hemorrhagic stroke that comprises 3% of all stroke types, 85% of which are caused by the rupture of intracranial aneurysms (IAs) (Go et al., 2014; Macdonald and Schweizer, 2017). SAH caused by ruptured IAs remarkably increases the chance of mortality and morbidity by 50% (32–67%) despite advances in management (Huang and van Gelder, 2002; van Gijn et al., 2007). Stroke has many genetic and environmental risk factors. A study of single-gene diseases has shown that common variations in approximately 35 loci are strongly associated with the risk of hemorrhagic stroke (Dichgans et al., 2019). In addition, various healthrelated and environmental factors, such as high blood pressure, diabetes, high cholesterol, high body mass index, smoking, and a history of hemorrhagic stroke, all increase the risk of hemorrhagic stroke (Donnan et al., 2008). The main causes of death are associated with the sharp increase in intracranial pressure due to initial hemorrhage and cerebral edema, which eventually lead to cerebral hernia. Several studies have shown that 20% of the fatality caused by SAH occurs because no medical attention is given, 30% of it occurs within 24 hours of onset, and 40–60% of the patients with early brain injury (EBI) caused by initial bleeding and delayed cerebral ischemia (DCI) resulting from cerebral vasospasm constitute the cause of subsequent mortality within a month, although emergency surgery and medication with drugs such as mannitol, Nimotop, and neurotrophin were performed after SAH (Hasegawa et al., 2011; Korja and Kaprio, 2016; Grasso et al., 2017). For survivors, long-term care is required in one-third of patients, and some recovered patients still suffer from neurological and/or cognitive deficits (van Dijk et al., 2016). Although there are marked improvements in microsurgical clipping and endovascular coiling treatments, the mortality of SAH is not greatly reduced because the residual blood in the subarachnoid space continues to stimulate and damage the brain cells. Therefore, the pathophysiological mechanism of SAH has been the focus of research in recent years. Previous experimental studies have discovered a series of pathological mechanisms following SAH, such as inflammation, apoptosis of nerve cells and vascular endothelial cells, and oxidative stress (OS) (Lucke-Wold et al., 2016; Mo et al., 2019). Many post-SAH responses, such as OS and inflammation, eventually lead to the death of neurons, vascular endothelial cells, and glial cells (Sekerdag et al., 2018). Recent experimental studies have shown that apoptosis is closely associated with cerebral injury after experimental SAH (Wu et al., 2020b). Apoptosis refers to the orderly death of cells under the autonomous control of genes. It is controlled by multiple genes, such as the bcl-2 family, Caspase family, and oncogenes, such as C-myC and tumor suppressor gene p53 (Fleisher, 1997). Several molecules and/or pathways, such as the phosphatidylinositol-3-kinase/AKT signaling pathway (Endo et al., 2006), Mas/PKA/CREB/UCP-2 pathway (Mo et al., 2019) and p53 (Ling et al., 2019), are activated after SAH, which may cause blood-brain barrier (BBB) dysfunction and neuronal apoptosis. Some anti-apoptotic proteins or drugs, such as Mas, AVE 0991, melatonin, and heat shock protein 22, can greatly improve neurological function after SAH (Bader et al., 2014; Shi et al., 2018; Mo et al., 2019; Fan et al., 2021). In this review, we aimed to understand the mechanism and relevant mediators related to neuronal apoptosis and potential therapeutic targets after SAH.
Retrieval Strategy
Literature review was electronically performed using PubMed database. The following combinations of key words were used to initially select the articles to be evaluated: apoptosis and subarachnoid hemorrhage, neuronal apoptosis and subarachnoid hemorrhage, cell death and subarachnoid hemorrhage, apoptosis and stroke, neuronal apoptosis and stroke, cell death and stroke, treatment and subarachnoid hemorrhage, targets and subarachnoid hemorrhage, stem cells and subarachnoid hemorrhage, stem cells and stroke. Most of the selected studies (80% of all references) were published from 2011 to 2021.
Apoptosis Pathways
Apoptosis can occur through three different pathways, namely, the extrinsic pathway, intrinsic pathway, and endoplasmic reticulum (ER) stress-induced pathway, depending on the site of apoptosis. In addition, other molecular mechanisms such as p53 and oxidative stress pathways are also associated with apoptosis after SAH (Hasegawa et al., 2011).
Extrinsic mechanis
mThe external apoptosis pathway plays an important role in promoting the regulation of cell apoptosis by death receptors on the cell surface without passing through the mitochondrial and stress ER-induced pathways. Death receptors, such as tumor necrosis factor (TNF) receptor, P2X7R, death receptor 4/5, and Fas, mediate the apoptotic pathway in hemorrhagic stroke by activating caspase-8 or -10 (Martin-Villalba et al., 1999; Rosenbaum et al., 2000). Once activated, caspase-8 can activate the downstream effects of caspases to produce tBid through direct proteolytic cleavage or indirectly through the cleavage of the BH3-only protein Bid, which is translocated to mitochondria and induces Bax activation and mitochondrial outer membrane permeability (Zhao et al., 2018a). TNF-α and Fas ligands can induce partial neuronal apoptosis during the inflammatory process. The apoptotic pathway of motor neurons is Fas-dependent, involving p38 and NO, resulting in classic caspase-dependent apoptosis (Haase et al., 2008). For example, the stimulation of P2X7R can activate caspase-1, which promotes the maturation and release of IL-1β and increases IL-1β concentrations, thus triggering the induction of TNF, which also has pro-apoptotic effects causing the expansion of cell apoptosis (Lee et al., 2016). Some evidence suggests that extrinsic apoptosis may play a causal role in neuronal death after stroke (Li et al., 2006b), but these models lack clear evidence that caspase-8 leads to death because caspase-8 deficiency (and FADD) is fatal to embryonic mice. However, Krajewska et al. (2011) solved this problem by using mice that specifically lacked caspase-8 expression in neuronal cell types and showed that neuron-specific caspase-8 loss makes neurons resistant toin vitro
TNFreceptor connection-induced apoptosis and leads to increased neuronal survival. Many previous experimental studies have shown that TNF-α and IL-1β expression is upregulated, and neuronal damage and apoptosis occur to varying degrees after SAH (Sekerdag et al., 2018; Guo et al., 2019; Lai and Du, 2019). Therefore, the extracellular apoptotic pathway plays an important role in neuronal apoptosis after SAH (Figure 1
).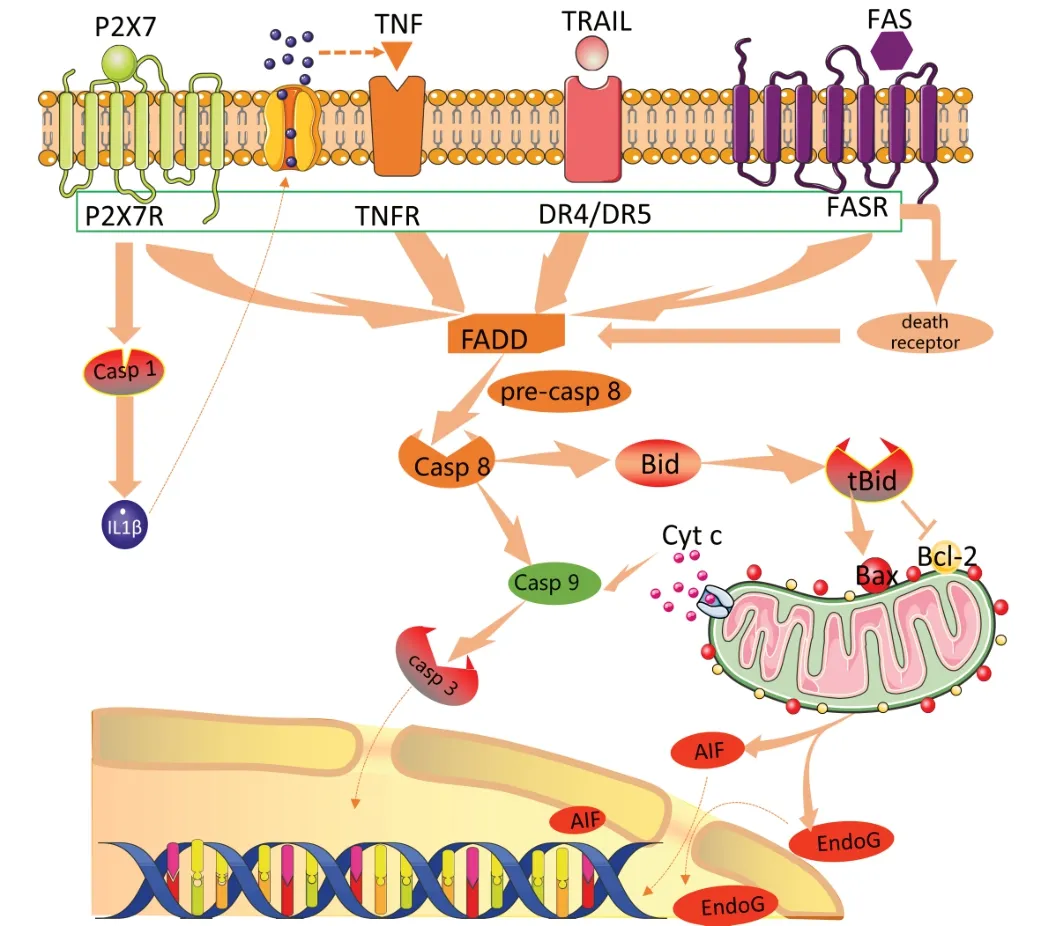
Figure 1|Extrinsic apoptosis pathway after SAH.Death receptors on the cell membrane, such as P2X7R, TNFR, DR4/DR5 and FSAR, can be stimulated by apoptotic signals to activate FADD, and then cause caspase-dependent apoptosis cascade reaction. AIF: Apoptosis inducing factor; DR4/DR5: death receptor 4/ death receptor 5; FADD: Fas-associating protein with a novel death domain; SAH: subarachnoid hemorrhage; TNF: tumor necrosis factor.
Mitochondrial (intrinsic) pathway
The mitochondrial pathway (also known as the intrinsic pathway) is mainly regulated by B-cell lymphoma-2 (Bcl-2) family proteins, which contain proapoptotic (e.g., Bax, Bak, Bad, Bid, Bim, and Noxa) and antiapoptotic (e.g., Bcl-2 and Bcl-xL) proteins (D’Orsi et al., 2017). Under normal circumstances, Bcl-2 and Bcl-xL, Bax and Bak form heterodimers, which jointly maintain the integrity of the mitochondrial outer membrane and prevent mitochondrial apoptosis (Edlich et al., 2011; Todt et al., 2015). Under apoptotic conditions, Bax and Bak are activated and aggregate at the mitochondrial outer membrane, where they are oligomerized and mediate mitochondrial outer membrane permeabilization, leading to the release of pro-apoptotic factors, such as cytopigment C (Lovell et al., 2008). In addition, Bax and BH3 proteins increase in expression and then combined with Bcl-2 and BclxL to release Bax/Bak. Free Bax and Bak form oligomers and are embedded in the outer membrane of mitochondria (Dlugosz et al., 2006; Youle and Strasser, 2008). Once the outer mitochondrial membrane permeability increases, mitochondrial proteins, such as cytochrome c, are released into the cytoplasm. Cytochrome c can interact with apoptotic protease activating factor-1 (Apaf1) to form apoptosomes and cause caspase-9 activation. Caspase-9, as an initiator of the cytochrome-dependent cascade, activates caspase-3 and causes DNA damage (Hasegawa et al., 2011). In addition, proteins such as AIF, SmaC, and Endo G are also associated with mitochondrial apoptosis. During apoptosis, these proteins are also released from the mitochondrial membrane space into the cytosol. SmaC OMI can bind to inhibitor of apoptosis proteins (IAPs) and resist the inhibition of IAPs on caspase 3 and caspase 9, which is a caspase-dependent protein. AIF and EndoG can translocate to the nucleus to cause chromatin condensation and large-scale DNA fragmentation, which are noncaspase-dependent proteins (Xiong et al., 2014;Figure 2
). Cleaved caspase-3 levels have been reported to be upregulated in the hippocampus and cortex after SAH (Zhang et al., 2019b). Mitochondrial dysfunction is a common cause of neuronal apoptosis in SAH (Wang et al., 2018), and it is considered to be a crucial therapeutic target for EBI after SAH (Mo et al., 2019). The activation of mitochondrial aldehyde dehydrogenase 2 (ALDH2) has been reported to markedly preserve mitochondrial function via PKCε phosphorylation, which has been shown to provide marked protection against apoptosis in the setting of cardiac and cerebral ischemia/reperfusion injury (Aldi et al., 2014; Wang et al., 2017b). TGR5 combines with INT-777 to attenuate neuronal apoptosis via the cAMP/PKCε/ALDH2 pathway after SAH (Zuo et al., 2019). The c-Jun N-terminal kinase signaling pathway may be independent of the p38 and NF-κB signaling pathways and is upregulated to promote apoptosis in the SAH model (Ling et al., 2019). Docosahexaenoic acid alleviates apoptosis following SAH by improving mitochondrial dynamics in EBI (Zhang et al., 2018).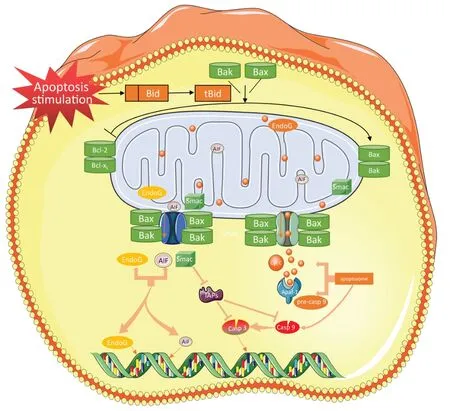
Figure 2|Mitochondrial pathway of neuronal apoptosis after subarachnoid hemorrhage.After stimulation of apoptosis signal, Bax and BH3 protein expression increased and Bax and BH3 combined with Bcl-2 and Bcl-XL to release Bax/Bak. Free Bax and Bak form polymeride and penetrate into the mitochondrial membrane to form ion channels and increase the permeability of the membrane, thus releasing Cyt C. Cyt C binds to Apaf-1 to recruit caspase-9 progenase to form apoptotic bodies and initiate downstream caspase cascade reactions. SMAC can combine with inhibitor of apoptosis proteins (IAPs) to resist the inhibition of caspase-3 and -9 by IAPs and promote apoptosis. AIF and Endo G released by mitochondria can be transposed to the nucleus, causing chromatin condensation and DNA fragmentation. AIF: Apoptosis inducing factor; Cyt C: cytochrome C; SMAC: second mitochondria-derived activator of caspases.
Endoplasmic reticulum pathway
The endoplasmic reticulum (ER) is the site in which secreted proteins and membrane proteins are synthesized and folded. Properly folded and modified proteins can be transported to the Golgi apparatus for further processing. In addition, the ER also stores Caand regulates Cametabolism. The imbalance of Caions in the endoplasmic reticulum and the increase in misfolded or unfolded proteins will cause endoplasmic reticulum stress (ERS) (Zeeshan et al., 2016). A moderate ERS response can reduce protein synthesis, ensure that proteins are folded correctly, and maintain intracellular Cahomeostasis, but an excessive stress response can trigger apoptosis signals and promote apoptosis. The unfolded protein response (UPR) is an important selfprotection mechanism of cells against ERS (Sun et al., 2017). There are three ER transmembrane proteins (Irel, ATF6, and PERK) in mammalian cells, all of which play a role in the aggregation of the UPR in the cavity (Ghemrawi and Khair, 2020). They regulate the quality and quantity of basic leucine zippers and produce different responses to different UPRs through interaction. If this response does not sufficiently reduce ERS, apoptosis may occur (Oslowski and Urano, 2011). The UPR response is a cellular protective response. However, whether this response can restore ER homeostasis depends on the intensity and duration of the stimulus. If the stimulation is too strong or lasts too long and these responses are not enough to restore and maintain ER homeostasis, programmed death will be initiated, leading to apoptosis (Sprenkle et al., 2017). The three apoptotic pathways are named IRE1α/c-Jun N-terminal kinase (JNK), PERK/eukaryotic promoter-2 (eIF2α)-CHOP, and ATF6-CHOP (Doyle et al., 2011; Xu et al., 2018a). Under normal cell operation, the ER mainly releases Cafrom the ER lumen into the cytoplasm through RyR and IP3R channels, and intracellular Cais pumped into the ER through a calcium pump, thus maintaining the ER Cahomeostatic state. When the ER receives the stress signal, Cahomeostasis in the ER is unbalanced, and a large amount of Caenters the cells and mitochondria. It affects the activity of mitochondria and Bcl-2 family proteins, leading to apoptosis. It also activates the caspase cascade and affects apoptosis (Burton et al., 2017; Marchi et al., 2018;Figure 3
). In recent years, an increasing number of experimental studies have supported the view that ERS plays an important role in neuronal apoptosis after SAH (Li et al., 2016a; Zhao et al., 2017). SAH activates ERS in a variety of ways to induce apoptosis. Upregulated expression of caspase-12, a proteolytic enzyme specific to the ER, was observed in the ER after experimental SAH, and its activation can be considered a marker of neuronal apoptosis mediated by ERS (Datta et al., 2018). The downstream molecule of ASK1, JNK, is a member of the signal transduction protein family, which regulates the expression of antiapoptotic genes by activating MAPKS, JNKs, and p38MAPKS, thereby inducing apoptosis (Li et al., 2006a). CHOP can mediate apoptosis by up-regulating the sensitivity to OS of cells and downregulating the secretion of the anti-apoptotic B-cell lymphoma-2 (Bcl-2) protein (Hetz, 2013; Qi et al., 2018). ERS can lead to calcium disorder in the ER, and inhibition of ER stress can restore the homeostasis of the ER (Han et al., 2019). ER-mediated apoptosis can exist in neurons and endothelial cells, resulting in the destruction of the BBB and irreversible apoptosis of neurons. A recent study has reported that the morphology of the coarser ER changed within 6 hours after SAH, the swelling of cortical neurons was the most severe at 24 hours and subsequently subsided within 24–48 hours (Tian et al., 2020). There was no marked difference between the SAH and normal groups at 72 hours, which was consistent with the expression of stress-related apoptotic protein cleavage of ER, caspase-12 ASK1, p-JNK in a rat model (Tian et al., 2020). PERK/eIF2/ATF4/CHOP signaling is activated after SAH and promotes apoptosis. Tauroursodeoxycholic acid inhibits this signaling pathway, reducing ER stress-induced apoptosis and SAH-associated cerebrovascular dysfunction (Chen et al., 2020b). The persistence of the UPR indicated that ERS was not relieved, and homeostasis was not restored. The severity and duration of ERS are related to the survival of neurons (Hetz and Saxena, 2017). Tauroursodeoxycholic acid inhibits the PERK/eIF2α/ATF4/CHOP signaling pathway and reduces ERS-mediated apoptosis, thereby improving SAHrelated cerebrovascular dysfunction (increased cerebral cortical perfusion and decreased BBB permeability) and neurological function (Chen et al., 2020b). Apelin-13 plays a neuroprotective role in EBI following SAH by inhibiting the ERS response pathway ATF6/CHOP (Xu et al., 2018a).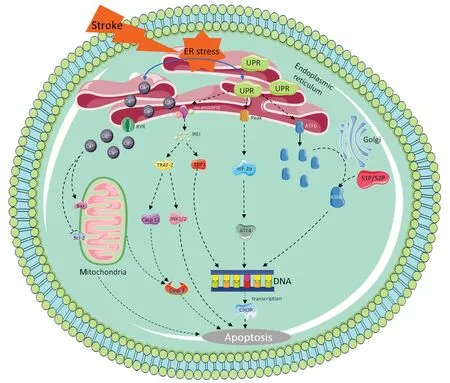
Figure 3|ER apoptotic pathway in neuronal apoptosis after subarachnoid hemorrhage.ER stress induced by subarachnoid hemorrhage activates three transmembrane proteins on the ER, and induces apoptosis through PEAK/EIF-2α/ATF4/CHOP, IER1 and ATF6/CHOP, respectively. Meanwhile, Ca2+ imbalance leads to a large amount of Ca2+ entering cytoplasm and mitochondria, which affects the activity of bcl-2 family proteins in mitochondria and leads to cell apoptosis. ER: Endoplasmic reticulum; UPR: unfolded protein response.
p53
p53, as a tumor suppressor gene, is also the most important factor in generating the apoptosis process in response to ischemia, hemorrhage, hypoxia, and severe DNA damage (Culmsee and Mattson, 2005; Chai et al., 2022). Under normal conditions, p53 levels are low and even undetectable. However, stress signals such as DNA damage and hypoxia can stabilize the p53 protein and induce cellular p53 levels to increase by post-translational modifications such as phosphorylation, ubiquitination, and acetylation (Zhou et al., 2005). Activation of p53 can cause a variety of responses, including cell cycle arrest or apoptosis (Wang et al., 2015b). p53 is one of the crucial factors in neuronal cell death. For example, it was upregulated at both 24 and 72 hours after SAH, and an inhibitor of p53 decreased brain injury and neuronal cell death (Gao et al., 2009). The Bcl-2 family is well known to participate in cell apoptosis and is mediated by p53 on the mitochondria, leading to apoptosis (Li et al., 2010). Moreover, the expression levels of p53 were markedly increased in basilar artery endothelial cells of rats post-SAH, and apoptosis was detected (Li et al., 2016b). In addition, it was also reported that pifithrin-α, a p53 inhibitor, suppresses p53 protein expression, decreases microRNA-22 expression and inhibits Bax protein expression in a SAH mouse model (Yu et al., 2018a). p53 mediates apoptosis mainly by activating mitochondrial and death receptor-induced apoptotic pathways, which induce caspase signaling and apoptosis (Yu and Zhang, 2005).Oxidative stress
OS refers to a state of imbalance between oxidation and antioxidation in the body. The balance tilts toward oxidation, resulting in increased secretion of protease and the production of a large number of oxidative intermediates (Deng et al., 2021). OS is a type of negative effect produced by free radicals in the body that can induce cell apoptosis and has an extensive association with apoptosis (Mo et al., 2019). It has been shown that hemoglobin (Hb) activates the caspase pathway and induces the apoptosis of cultured cortical neurons and microvascular endothelial cells (Katsu et al., 2010). After SAH, the central nervous system is exposed to high levels of Hb and Hb degradation products in the subarachnoid space. This pathological process produces excessive ROS and RNS and promotes cerebral vasospasm, cerebral stenosis, and DCI (Vergouwen et al., 2010). In addition, a number of harmful events occur in SAH survivors, including altered ion homeostasis, excitatory toxicity, disruption of vascular integrity, OS, inflammation, apoptosis, autophagy, and the activation of NOS pathways (Fan et al., 2017; Han et al., 2017a; Shi et al., 2017c; Zhang et al., 2017).
Apoptosis in Early Brain Injury
EBI occurs within 72 hours of IA rupture and is considered to be a major factor leading to adverse outcomes (Shi et al., 2017b). EBI emphasizes immediate global brain injury caused by temporarily increased intracranial pressure and reduced cerebral blood flow after SAH (Conzen et al., 2019). The initial blood load causes an increase in the intracranial pressure, which has been demonstrated by endovascular filament perforated animal models. Intracranial pressure increased to 40 mmHg immediately after SAH in this model and then dropped to a plateau (15–25 mmHg), while cerebral perfusion pressure dropped from 70 mmHg to 35–40 mmHg, and cerebral blood flow decreased by 20–30% from baseline before SAH (Tӧrӧk et al., 2009). The potential mechanisms of EBI include neuronal apoptosis, neuroinflammation, OS, and BBB destruction (Zhu et al., 2018). Transient systemic ischemia and local blood infiltration after SAH can inhibit oxidative phosphorylation and cause mitochondrial homeostasis disorder and reactive oxygen species storms, which are harmful to the vitality and development of neurons. Over the past few decades, mitochondria have been recognized as major regulators of neural function and neuronal activity under SAH, especially EBI (Mo et al., 2019; Zhang et al., 2019a). Mitochondria are not only the main sites of ATP production but are also involved in the formation of reactive oxygen species and cell apoptosis. Importantly, there is growing evidence that mitochondrial biogenesis and fusion/fission play a critical role in maintaining mitochondrial function and homeostasis (Sekerdag et al., 2018). Excessive mitochondrial division will disrupt mitochondrial homeostasis, leading to the production of pan-ROS (Wang et al., 2017a) and the transfer of cytochrome C to the cytoplasm, thereby triggering apoptosis (Du et al., 2019). In addition, an increasing number of experimental studies support the view that ERS plays an important role in the pathophysiological process after SAH (Yan et al., 2014; Zhao et al., 2017). Overactivation of ERS causes calcium release and OS, which further triggers downstream cascades that lead to inflammation and apoptosis (Sprenkle et al., 2017). Li et al. (2015a, b) showed that activation of the inflammatory proteome containing the three proteins (NLRP3) in the thioredoxin interacting protein (TXNIP)/NODlike receptor pyrin domain can link ER stress to brain tissue inflammation and apoptosis. One of the main pathophysiological mechanisms contributing to EBI development is the activation of the apoptotic pathway, and antiapoptotic treatments are effective therapeutic strategies against EBI (Shi et al., 2017a; Zhao et al., 2018b). Apoptosis may be seen in cortical, subcortical, or hippocampal neurons and the endothelium after SAH (Yuksel et al., 2012). After SAH, a large amount of blood enters the subarachnoid space, and erythrocyte lysates release Hb and other cell contents covering the surface of neuronal cells, glial cells, and other brain cells. Iron released from Hb induces apoptosis through lipid peroxidation, which then induces extensive apoptosis in cortical, subcortical, and hippocampal neurons and leads to high mortality and disability rates (Sekerdag et al., 2018).
Apoptosis in Delayed Cerebral Ischemia and Cerebral Vasospasm
Once past the EBI stage of SAH, prognosis often depends on the occurrence of DCI, which is detected in approximately 40% of patients by cerebral tomodensitometry and 80% of patients with brain magnetic resonance imaging (El Amki et al., 2018). Finally, the prognosis of patients with SAH is usually unfavorable, with mortality estimated in clinical investigation ranging from 20% to 60% (Bogason et al., 2014) and neuropsychological disorders observed in 50–60% of patients who survived (Mayer et al., 2002). Recent clinical studies have confirmed that cerebral vasospasm is one of the factors contributing to DCI (Francoeur et al., 2022; Labak et al., 2022). It may occur 3–14 days after SAH and is visible by computed tomography angiography in up to 70% of patients (Yuksel et al., 2012). In SAH animal experiments, by reducing basilar artery apoptosis at 24 and 72 hours, we were able to greatly prevent severe vasospasm by measuring the diameter of the basilar artery. Although the time course of apoptosis in this model was 24 hours, we found that the apoptosis mechanism was significantly upregulated at 72 hours by western blotting (Cahill et al., 2006). In addition, microcirculation spasm, microthrombogenesis, extensive cortical depolarization, and brain dysfunction are also considered to be important reasons for the development of DCI (Geraghty and Testai, 2017). Many pathological processes have been suggested as possible mechanisms for delayed cerebral vasospasm after SAH, including endothelial injury, smooth muscle contraction, vascular reactivity changes, and inflammation and/or immune responses to vascular walls. Apoptosis exists in vascular tissues with different degrees of necrosis after SAH, which is involved in the proliferation of spasmodic arterial smooth muscle cells (Tsai et al., 2020). The cascade of apoptosis may be the cause of vasospasm (El Amki et al., 2018). Large vessel vasospasm leads to cerebral ischemia after SAH. Blood cell contents are released into the subarachnoid space, inducing the production of a large number of inflammatory factors, leading to neuroinflammation and loss of BBB function and causing apoptosis of neurons and endothelial cells. Persistent and irreversible damage ultimately leads to DCI and a poor prognosis. Apoptosis is considered to be one of the most critical factors that may be connected with delayed neurological deterioration and poor long-term prognosis (Chen et al., 2014).
Blood-Brain Barrier Dysfunction and Cerebral Edema Facilitate Apoptosis
The BBB is made up of endothelial cells, pericytes, basement membranes, and astrocyte end feet. The properties of the BBB are largely manifested within ECs but are induced and maintained by critical interactions with mural cells, immune cells, glial cells, and neural cells, which interact in the neurovascular unit (Daneman and Prat, 2015). The BBB, with low vascular permeability, limits the entry of potentially neurotoxic plasma components, blood cells, and pathogens into the central nervous system (Won et al., 2011). Many factors are unique to VCECs forming the BBB, including endothelial tight junctions and adherens junction proteins, bulk-flow transcytosis, pinocytosis, nonselective fenestrae, and the suppression of leukocyte adhesion molecules (Obermeier et al., 2013). Many diseases, such as hemorrhagic stroke and ischemic stroke, cause BBB dysfunction. It has been reported that BBB disruption occurs as early as 10 minutes after ictus and can persist up to 7 days (Tso and Macdonald, 2014). In addition, a great amount of blood flowing into the subarachnoid space leads to acute intracranial hypertension, and subsequent release of blood content promotes apoptosis of brain cells (neurons, microglia, astrocytes, endothelial cells, and pericytes;Figure 4
).
Figure 4|Blood enters the subarachnoid space after SAH.A large number of blood contents (erythrocyte, leucocyte, platelet) enter the subarachnoid space after SAH. Erythrocytes decompose into oxygenated hemoglobin, leading to deterioration of BBB (endothelial cell, pericyte, astrocyte, basement membrane) and neuronal apoptosis. BBB: Blood-brain barrier; SAH: subarachnoid hemorrhage.
Destruction of pericyte cells results in increased BBB permeability
Pericytes surround endothelial cells and are located in the basement membrane. Under physiological conditions, pericytes can regulate BBB integrity, microvascular remodeling and cerebral blood flow, maintain vascular structure stability, remove toxic metabolites in the central nervous system, and mediate neuroinflammation (Liebner et al., 2011). Peripheral brain cells, whose coverage rate in the central nervous system is higher than that of peripheral blood vessels, are necessary for the formation of the BBB during development, and their coverage rate affects BBB permeability. Under pathological conditions, TNF, one of the mediators of neuroinflammation, can stimulate the secretion of matrix metalloproteinase 9 by pericytes, which leads to basement membrane rupture, pericyte cell migration, and BBB injury (Takata et al., 2011). Currently, pericyte markers include platelet-derived growth factor receptor β, NG2 proteoglycan, and α-smooth muscle actin. A recent study has reported that inflammatory mediators can alter pericyte function, and exposure of pericytes to TNF or IL-1β leads to downregulation of pericyte markers (platelet-derived growth factor receptor, NG2 proteoglycan, and α-smooth muscle actin), loss of basic pericyte function, and BBB damage (Persidsky et al., 2016). Moreover, soluble platelet-derived growth factor receptor β was elevated in the cerebrospinal fluid after SAH and subsequently promoted BBB injury (Liu et al., 2018a).
Cerebral vascular endothelial cells damage
Under physiological conditions, cerebral vascular endothelial cells (CVECs) maintain the integrity of the BBB, regulate the formation of thromboses, and regulate vascular tones. However, early events after aSAH trigger CVEC dysfunction and apoptosis. A large number of erythrocytes, white blood cells and other blood contents enter the brain. Cytolysis products, especially oxyhemoglobin, induce apoptosis of CVECs via caspase-8 or -9 (Peeyush Kumar et al., 2019). In 2000, Zubkov et al. first discovered that typical endothelial cell apoptosis secondary to cerebral vasospasm was induced by SAH. CVEC apoptosis is reported to occur 24 hours after aSAH (Friedrich et al., 2012). Apoptosis markers, such as cleaved caspase-3 and TUNEL staining, colocalized with endothelial staining 10 minutes after aSAH in rat models. The endothelia of parenchymal vessels were destroyed and detached from the basal lamina within 10 minutes (Friedrich et al., 2010, 2012). Furthermore, postmortem human studies reported that CVEC death after aSAH was mediated via OxyHb, which elevates intracellular Caand matrix metalloproteinase 9 levels (Guo et al., 2015; Peeyush Kumar et al., 2019).
Vasogenic and cytotoxic edema lead to cerebral edema
Early cytotoxicity and vasogenic edema occurred within 72 hours after SAH (Weimer et al., 2017). To confirm the presence of edema after SAH injury, magnetic resonance imaging (MRI), particularly T2WI imaging (T2WI), was used to provide visual information on BBB rupture and tissue edema (Jadhav et al., 2008). Generally, the T2 relaxation time in T2WI sequences is considered to be a valuable parameter reflecting hydrodynamics and is sensitive to water binding, and the T2 time is longer in the SAH group than in the normal group, suggesting the presence of vasogenic edema. Further Evans blue tests and IgG staining also confirmed the destruction of BBB integrity. Therefore, we can conclude that the development of vasogenic edema increases BBB destruction and the subsequent accumulation of cerebral edema. Cytotoxic edema is another type of brain edema in addition to vasogenic edema. Diffusion weighted imaging and apparent diffusion coefficient imaging are more sensitive and noninvasive tools for detecting intracerebral cytotoxic edema (Jadhav et al., 2008). A large amount of blood flows from the blood vessels to the subarachnoid space after SAH, which leads to a sharp increase in intracranial pressure, reducing cerebral blood flow and cerebral perfusion pressure and thereby causing cerebral cytotoxic edema (Weimer et al., 2017). The pathogenesis of cytotoxic brain edema is mainly related to the decline in sodium pump function. After SAH, acute hypoxia and anoxia may reduce the production of ATP, resulting in decreased activity of the sodium pump, which depends on ATP to provide energy, so Nacannot be transported actively to the outside of the cell, and water enters the cell to restore the balance, resulting in excessive Naand water accumulation in brain cells (Bano et al., 2005). In addition, the imbalance of Cahomeostasis is also an important reason for brain edema. Under normal conditions, the extracellular Caconcentration is 10,000 times higher than the intracellular Caconcentration, and such a large concentration difference is completely maintained by the Capump. Cerebral edema after SAH causes ischemia and hypoxia, the Capump is imbalanced, and Caenter the cells, further aggravating cerebral edema (Azad et al., 2016; Boyacı et al., 2019).
Treatment
Novel molecular and cellular treatment strategies
SAH in 85% cases is caused by ruptured intracranial aneurysms (Macdonald and Schweizer, 2017). Therefore, once SAH is detected by examination, treatment should first identify the cause of hemorrhage. If SAH is caused by ruptured IAs, the ruptured IAs should be treated first. Cerebral edema and cerebral vasospasm caused by SAH should be alleviated at the same time (Maher et al., 2020). In the long term, current approaches seek to minimize damage to neurons while maximizing the repair potential of the lost neurons (Yuksel et al., 2012). Because damaged neurons are difficult to regenerate, treatment is often insufficient to restore physiological function. To inhibit the components of post-SAH apoptosis cascade, some potential molecular therapies may include mitochondrial apoptosis pathway inhibitors, ERS inhibitors, ROS inhibitors, p53 inhibitors, Caantagonists, and apoptosis pathway inhibitors (Cahill et al., 2006; Wang et al., 2015b; He et al., 2016;Figure 5
). In recent years, therapies targeting potential receptors, signaling pathways, and miRNAs have received increasing scientific attention. The AKT signaling pathway has been widely reported to be involved in the pathophysiological mechanism after SAH, and the activation of AKT contributes to the reduction of EBI and neuronal apoptosis after SAH (Hasegawa et al., 2011). For example, KP54 can reduce neuronal apoptosis and oxidative stress after SAH and improve neurobehavioral deficits in rats by activating the AKT signaling pathway (Huang et al., 2021). 5-Lipoxygenase inhibitors attenuate neuronal apoptosis and inflammation through the AKT signaling pathway after SAH (Liu et al., 2021). C-Abl tyrosine kinase mediates neuronal apoptosis after SAH by activating the Akt/GSK3β pathway (Yan et al., 2021). AKT activation (phosphorylation) can be activated by recombinant OX40. Silencing of tenascin-C, FGF-2, and apelin-13 can markedly reduce neuronal apoptosis and neuroinflammation induced by SAH (Liu et al., 2019; Okada et al., 2019; Tong et al., 2020; Wu et al., 2020b). Caspase 3 inhibitors, such as liraglutide (Tu et al., 2021), calpeptin (Zhou and Cai, 2019), and methazolamide (Li et al., 2016c), can inhibit the expression of caspase-3, thereby reducing the cascade of apoptosis induced by caspase 3 and ultimately reducing neuronal apoptosis.Table 1
lists several potential targets for SAH recovery. Next to targeting apoptosis inhibitors, focus can be led on the antioxidant drugs. Nanomaterials loaded with antioxidant drugs are emerging in recent years. Astaxanthin, for example, is an antioxidant that has been hampered by its easy degradation and low bioavailability as a therapeutic agent for clinical advancement. The stability and solubility of astaxanthin are greatly increased by nanomaterial encapsulation.In vitro
andin vivo
studies have confirmed that it can inhibit neuronal apoptosis after SAH through an antioxidant effect (You et al., 2019; Cai et al., 2021). NLRP3 inflammasomes are involved in neuroinflammation and apoptosis after SAH (Xu et al., 2021). Regulation of NLRP3 inflammasome activation at the molecular level may contribute to development of potential new therapeutic approaches (Bai et al., 2021). The inflammasome inhibitor has revealed a neuroprotective effect. In addition, miRNAs, as endogenous noncoding short single-stranded RNAs that regulate gene expression at the post-transcriptional level, are also gradually becoming new molecular targets after SAH. While therapies targeting individual genes have not been successful due to complex overlapping pathways, miRNAs are particularly useful for their ability to simultaneously regulate multiple target genes. Mice overexpressing miR-132 were less likely to develop nervous system defects (Sekerdag et al., 2018). Lentiviral overexpression of miR-126 has a protective effect against ICH and exerts an antiapoptotic effect by downregulating caspase-3 levels (Kong et al., 2017). Moreover, miR-103-3p was significantly upregulated in experimental models of SAH. MiR-103-3p plays a neuroprotective role in reducing neuronal death by reducing caveolin-1 (Xu et al., 2018b). Experimental models of SAH showed that use of apoptosis-related inhibitors provided some protection, but apoptosis still occurred (Yuksel et al., 2012). This may be due to a series of complex reactions after SAH, including apoptosis, inflammation, oxidative stress, autophagy, and coke death (Castro et al., 2018; Sekerdag et al., 2018; Wu et al., 2021). The efficacy of a single inhibitor is very limited, and the search for multi-target drugs will be a new direction of future research.
Table 1 |Potential neuroprotective targets for brain injury and neuron loss after subarachnoid hemorrhage

Figure 5|Potential targets for drug therapy after SAH.Several apoptosis pathways are activated after SAH, including extrinsic, mitochondrial, ER, p53 and ROS pathways. SAH can be treated with a variety of apoptosis inhibitors including death receptor inhibitors, mitochondrial apoptosis pathway inhibitors, ERS inhibitors, ROS inhibitors, p53 inhibitors, and Ca2+ antagonists. ER/ERS: Endoplasmic reticulum/endoplasmic reticulum stress; ROS: reactive oxygen species; SAH: subarachnoid hemorrhage.
Stem cell therapies
There was a study on nerve regeneration in mice after SAH treatment with stem cells as early as 2003 (Mino et al., 2003). The proliferation of neural progenitor cells after SAH was detected by adding 5-bromodeoxyuridine, a specific marker of cell proliferation. Mino et al. (2003) found that most 5-bromodeoxyuridine-positive cells became NeuN positive cells 30 days after SAH, indicating that the precursor cells had differentiated into mature neurons and promoted neurogenesis in the hippocampus. In addition, activation of neural progenitor cells after SAH may also occur in the adult brain. Sgubin et al. (2007) collected and analyzed brain tissue samples of patients with SAH and found SOX2 and Musashi mRNAs, two markers of proliferation of neural progenitor cells. Previous experimental studies have found that formation of new brain cells from neural stem cells can occur throughout life (Kumar et al., 2019). Neural stem cells, mainly located in the subventricular region and the subgranular region of the dentate gyrus, can proliferate and/or migrate to the damaged site to compensate and/or replace the lost neurons (Sekerdag et al., 2018; Li et al., 2022). Because there is no BBB, endogenous neurogenic stimulators released from the brain after SAH can be detected in cerebrospinal fluid, indirectly confirming the presence of neurogenesis (Kumar et al., 2019). Neurogenesis is characterized by the release of various neurotrophic factors, such as vascular endothelial growth factor, erythropoietin, and brain-derived neurotrophic factor (Wang et al., 2015a). However, generating sufficient numbers of neural stem cells for transplantation is a huge challenge. Therefore, many experimental studies have been forced to shift from neural stem cells to other stem cell sources. Mesenchymal stem cells can be derived from adipose tissue, bone marrow, dental pulp, and human umbilical cords. At the same time, it has the ability of multidirectional differentiation, which can induce nerve cells, muscle, adipose, and liver cells. Most importantly, it can penetrate the BBB, migrate, proliferate, and differentiate into nerve cells and is an ideal seed cell for nerve repair after SAH (Mukai et al., 2018; Song and Zhang, 2020). By injecting labeled mesenchymal stem cells into mice with SAH through the caudal vein, it was found that mesenchymal stem cells differentiated into nerve cells, reduced neuronal apoptosis, and improved nerve function in 14 days (Khalili et al., 2012). Stem cell therapy should be performed as early as possible, which can help reduce the subsequent damage caused by EBI. In recent years, treatment of subarachnoid hemorrhage with stem cells has developed rapidly. However, stem cell therapy for SAH has its limitations and challenges. First of all, mammalian brains may not be sufficiently or efficiently able to produce functional neurons. Secondly, the specific mechanisms of action and activation principle of stem cells are not clear, and research efforts are still needed. Moreover, most of the current researches are limited to preclinical studies, and the safety and reliability of stem cell therapy are not clear, especially the risk of tumor formation. Finally, detailed treatment strategies, including the optimal time window for treatment, stem cell types and numbers, delivery sites and pathways, need to be further explored.
Conclusion
In recent years, many pathophysiological mechanisms have been proposed for brain injury after SAH. Apoptosis is a very important link, including damage to neurovascular units, which leads to destruction of the BBB and the occurrence of cerebral edema. The stimulation of blood contents and secondary cerebral ischemia increase the apoptosis of brain cells and aggravate the neurological deficit of patients with SAH. Fortunately, a number of anti-apoptosis drugs and stem cell therapies are being developed and have been shown to be effectivein vivo
andin vitro
. However, many aspects need to be perfected before these approaches can be translated into clinical research. In this study, the pathophysiological mechanism and treatment methods related to apoptosis after SAH are discussed, which will provide direction for clinical treatment and development of targeted drugs in the future.Author contributions:
QT wrote the original draft. SL, SMH, WZ, and XYQ retrieved relevant literature and created table. JHC, CLL, and YJG created the figures. MCL reviewed and revised articles for final versions. All authors read and approved the final manuscript.
Conflicts of interest:
The authors declare that they have no competing interests.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- c-Abl kinase at the crossroads of healthy synaptic remodeling and synaptic dysfunction in neurodegenerative diseases
- Microglia depletion as a therapeutic strategy: friend or foe in multiple sclerosis models?
- Brain and spinal cord trauma: what we know about the therapeutic potential of insulin growth factor 1 gene therapy
- Functions and mechanisms of cytosolic phospholipase A2 in central nervous system trauma
- Cre-recombinase systems for induction of neuronspecific knockout models: a guide for biomedical researchers
- Prenatal programing of motivated behaviors: can innate immunity prime behavior?