综合生物信息学方法识别银屑病免疫抑制相关的潜在生物标志物
2022-06-23陈东宇杨晓雨王红心樊文龙刘兴华林泳煌何玉清
陈东宇,杨晓雨,王红心,樊文龙,刘兴华,林泳煌,何玉清
(1.东莞市寮步医院皮肤科,广东 东莞 523400; 2.广东医科大学 a.公共卫生学院流行病与卫生统计学系,b.医学系统生物学研究所,广东 东莞 523808)
银屑病是一种全身性免疫炎症疾病,典型的皮肤表现包括红斑、硬化和鳞屑斑块,其发病同时受到遗传易感性、环境危险因素、代谢、感染和生活方式等因素相互作用的影响[1]。银屑病可在任何年龄发病,估计影响着全世界1.25亿人[2]。大量基于人群的研究表明,银屑病在欧洲白人中的发病率最高,其次是黑人和西班牙裔人[3]。在银屑病异常的免疫系统中,角质形成细胞、黑色素细胞、树突状细胞、T细胞之间通过释放炎症细胞因子和趋化因子导致免疫细胞显著浸润,最终引起表皮过度增殖和角质形成细胞异常分化[4]。目前研究认为,银屑病潜在的病理机制涉及免疫细胞释放促炎性细胞因子增多,免疫细胞与角质形成细胞之间的病理串扰以及先天免疫系统与获得性免疫系统慢性激活的复杂相互作用[5]。识别这些遗传关联内在的发病分子机制是目前研究银屑病的研究热点。本研究运用生物信息学等方法筛选银屑病的保护基因,为明确其发病机制和临床治疗提供新思路。
1 资料与方法
1.1差异表达基因(differentially expressed genes,DEGs)的筛选 利用美国国家生物技术信息中心的基因表达数据库GEO(Gene Expression Omnibus)(https://www.ncbi.nlm.nih.gov/geo)进行基因表达谱芯片筛选及数据处理。利用R语言limma包对芯片数据进行标准化和DEGs的筛选,|log2FoldChange|≥1和校正后的P<0.05为差异有统计学意义。
1.2基因集富集分析(gene set enrichment analysis,GSEA) 采用clusterProfiler包对所有基因集合进行GSEA,参考基因集来自MSigDB数据库(http://www.gsea-msigdb.org/gsea/msigdb/index.jsp)的c2.cp.kegg.v7.4.entrez基因集。以P<0.05和错误发现率(false discovery rate,FDA)<0.25(矫正方法为Bonferroni矫正法)作为明显富集基因集的判定标准,取交集的通路进行分析。
1.3加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)构建 根据标准化后的基因表达矩阵,利用WGCNA包和limma包构建WGCNA,分别进行样本聚类,计算软阈值,构建无尺度网络和拓扑重叠矩阵。通过基因显著性和模块显著性评价基因/模块和临床症状信息之间的关联性,筛选出相关性最高的基因模块。
1.4DEGs的验证 根据WGCNA结果,计算各个模块对应的模块显著性和基因显著性,筛选与银屑病相关度最高的模块基因,绘制韦恩图得到交集DEGs。基于clusterProfiler包进行基因本体(Gene Ontology,GO)和京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)的富集分析以及可视化。基于最大集团中心性模式(maximal clique centrality,MCC)筛选连接度最高的前10个DEGs,并验证上述DEGs的表达情况。
1.5免疫细胞浸润评价及相关性分析 CIBERSORT算法利用RNA表达数据推断混合细胞群体中成员细胞类型的丰度[6]。加载R package e1071作为前置条件,模拟次数设为100,筛选出P<0.05的样本。分析22种免疫细胞在所有样本中的比例,查看数据集中各免疫细胞浸润的分布情况。绘制小提琴图来可视化免疫细胞浸润的差异,最后对DEGs和浸润免疫细胞进行Spearman相关分析。
2 结 果
2.1DEGs的识别 筛选得到GSE13355和GSE14905,两芯片均基于同一平台GPL570。选取GSE13355中的116个样本(58例银屑病患者皮损和58例非皮损皮肤样本)和GSE14905的61个样本(33例银屑病患者皮损和28例非皮损皮肤样本)进行分析。GSE13355芯片中共筛选出328个DEGs,其中上调基因176个、下调基因152个,见图1a。在GSE14905芯片中共筛选出1 171个DEGs,其中上调基因617个、下调基因554个,见图1b。
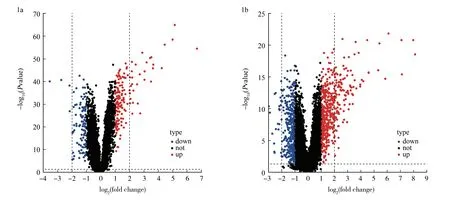
注:红点代表上调的差异表达基因,蓝点代表下调的差异表达基因
2.2GSEA 标准化处理GSE13355和GSE14905矩阵数据,以KEGG通路基因集为分类标准进行GSEA。获得的6条交集通路(嘧啶代谢、利什曼原虫感染、亚油酸代谢、细胞因子受体相互作用、趋化因子信号通路和嘌呤代谢)均受到抑制,见图2。

注:Rank in Ordered Dataset为在有序数据集中排名,Running Enrichment Score为富集得分,Ranked List Metric为列表指标排名
2.3WGCNA 计算得到GSE13355软阈值β=6,基于软阈值分步构建基因模块(图3a、3b)和共表达矩阵网络,并通过动态混合剪切得到13个以热图呈现基因模块与临床数据的相关性的结果(图3c),选择与银屑病皮损最显著相关的turquoise模块进行后续分析。该模块包括3 308个基因,该模块的模块基因与银屑病的基因显著性高度相关(r=0.97,P<0.001),见图3d。

注:Scale independence为尺度独立性,Scale Free Topology Model Fit signed R2为无尺度网络拟合指数R2,Soft threshold(power)为软阈值,Mean connectivity为平均连接度;Gene Dendrogram and module colors为基因树状图与模块颜色,Dynamic Tree Cut为动态聚类,Merged Dynamics为合并聚类;Module-trait relationships为模块-特性关系;Gene significance for psoriasis为银屑病的基因显著性,Module membership in turquoise module为turquoise模块中的模块基因
计算GSE14905软阈值β=18,基于软阈值分步构建基因模块(图4a、4b),得到8个以热图呈现基因模块与临床数据的相关性的结果(图4c),选择与银屑病皮损最显著相关的lightgreen模块进行后续分析。该模块包括986个基因,该模块的模块基因与银屑病的基因显著性高度相关(r=0.93,P<0.001),见图4d。

注:Scale independence为尺度独立性,Scale Free Topology Model Fit signed R2为无尺度网络拟合指数R2,Soft threshold(power)为软阈值,Mean connectivity为平均连接度;Gene Dendrogram and module colors为基因树状图与模块颜色,Dynamic Tree Cut为动态聚类,Merged Dynamics为合并聚类;Module-trait relationships为模块-特性关系;Gene significance for psoriasis为银屑病的基因显著性,Module membership in lightgreen module为lightgreen模块中的模块基因
2.4DEGs的筛选及功能富集分析 取交集得到89个交集的DEGs(图5a)。基于MCC模式获得连接度最高的前10个DEGs:CC趋化因子配体(CC motif chemokine ligand,CCL)20、CXC趋化因子受体4、S100钙结合蛋白(S100 calcium binding protein,S100)A7、CXC基序趋化因子配体2、角蛋白16、肽酶抑制剂3、E-选择素、S100A9、CCL8和富含脯氨酸的小蛋白2B(图5b)。

图5 交集差异表达基因的筛选 5a为GSE13355、GSE14905、turquoise模块和lightgreen模块交集的差异表达基因,5b为基于最大集团中心性模式的蛋白互作网络构建与差异表达基因的筛选
对DEGs进行GO功能注释(图6a),这些DEGs在生物过程方面主要富集在对细菌分子的反应、抗菌体液反应等生物学功能上;在分子功能方面主要富集在晚期糖基化终末产物受体受体结合钙依赖性蛋白结合细胞因子活性、Toll样受体4结合等生物学功能上;KEGG主要涉及3个通路:白细胞介素(interleukin,IL)-17信号通路、病毒蛋白与细胞因子及细胞因子受体的相互作用和昼夜节律(图6b)。
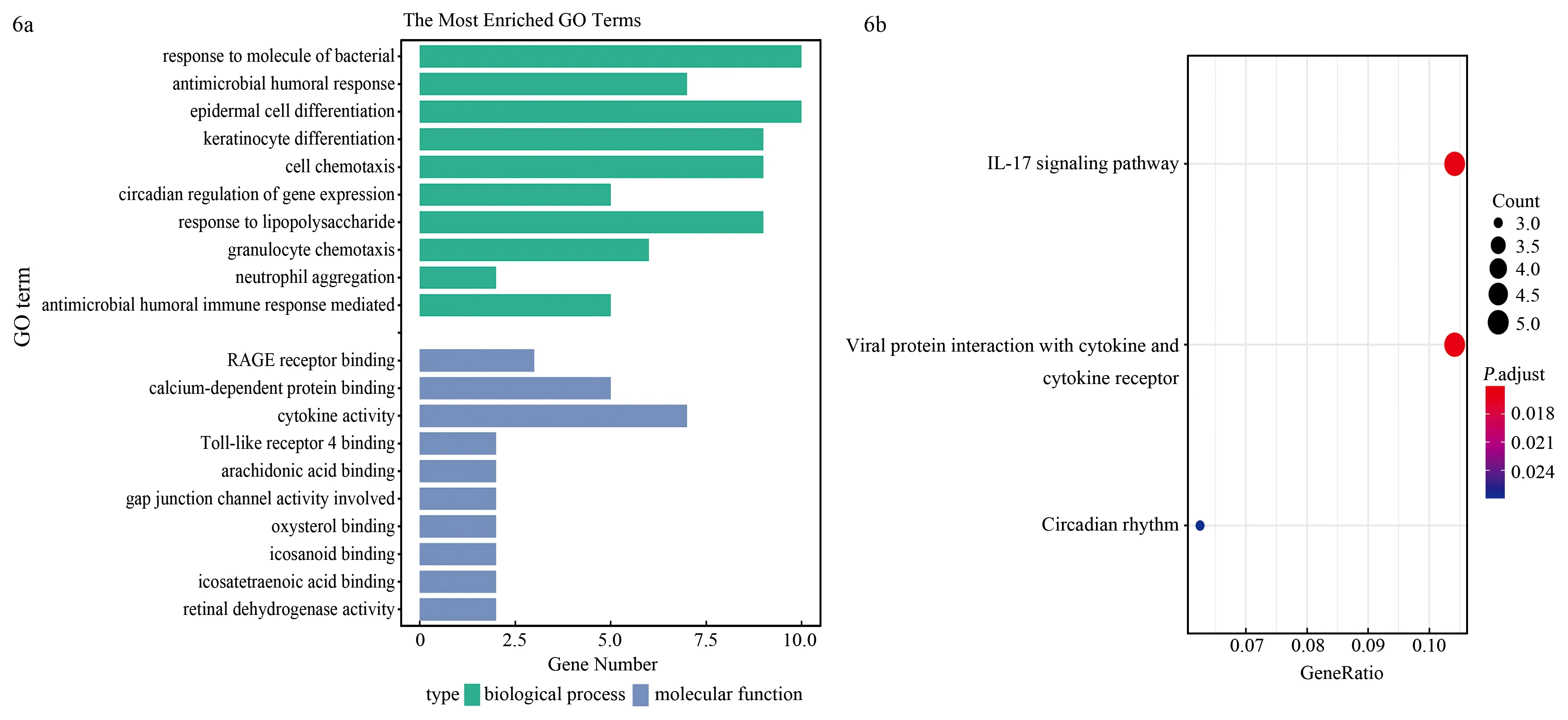
注:DEGs为差异表达基因;GO为基因本体,KEGG为京都基因和基因组百科全书,The Most Enriched GO Terms为最富集的GO条目,GeneRatio为基因比率
3a为不同软阈值对应的无尺度网络拟合指数和不同软阈值对应的平均连通性(β=6),3b为模块特征基因与临床特征相关性的热图,3c为基因聚类树及模块划分,3d为turquoise模块特征基因的散点图
2.5验证DEGs的表达情况 GSE166388包含 4例中国银屑病患者皮损和4例非皮损皮肤样本。验证上述89个交集DEGs在GSE166388的表达水平。结果显示,在银屑病患者皮损与非皮损组织间25个DEGs[S100A9、肽酶抑制剂3、醛脱氢酶1家族成员A3、细胞色素C氧化酶2、排斥性导向分子B(repulsive guidance molecule BMP co-receptor B,RGMB)、神经前体分化调节同系物(maturin,neural progenitor differentiation regulator homolog,MTURN)、核酸结合蛋白1、乳铁蛋白、LOC100506990、B细胞淋巴瘤2相关蛋白A1、锌指蛋白273(zinc finger protein 273,ZNF273)、F-box蛋白6、原肠(胚)形成糖蛋白(proteoglycan like sulfated glycoprotein,PAPLN)、S100A7、丙氨酸转氨酶2、CCL20、盐诱导激酶1(salt inducible kinase 1,SIK1)、尿苷磷酸化酶1、维甲酸相关孤儿受体 α(retinoic acid-related orphan receptor α,RORα)、单磷酸胞嘧啶-N-乙酰神经氨酸羟化酶假定蛋白(cytidine monophospho-N-acetylneuraminic acid hydroxylase,pseudogene,CMAHP)、Wnt家族成员2B(Wnt family member 2B,WNT2B)、蛋白酪氨酸磷酸酶非受体21(protein tyrosine phosphatase non-receptor type 21,PTPN21)、CCL8、S100A2和羟类固醇17β脱氢酶2]比较差异有统计学意义(P<0.05)。
2.6免疫细胞浸润及相关性分析 综合两个数据集,用箱线图表示22种免疫细胞在所有样本中的比例。各免疫细胞浸润的分布情况见图7a。含量前三的免疫细胞是活化的CD4记忆T细胞、未活化的树突状细胞和活化的肥大细胞。小提琴图显示,与银屑病非皮损组织相比,皮损组织的活化的CD4记忆T细胞、未活化的自然杀伤(natural killer,NK)细胞、活化的NK细胞、M1型巨噬细胞、未活化的肥大细胞和活化的肥大细胞的免疫浸润表达差异有统计学意义(P<0.001),见图7b。
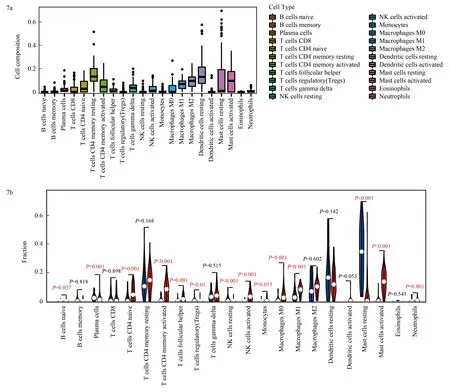
注:TME Cell composition为肿瘤微环境细胞组成,Cell composition为细胞组成,Cell Type为细胞类型;B cells naive为幼稚B细胞,B cells memory为记忆B细胞,Plasma cells为浆细胞,T cells CD8为CD8 T细胞,T cells CD4 naive为幼稚CD4 T细胞,T cells CD4 memory resting为未活化的CD4记忆T细胞,T cells CD4 memory activated为活化的CD4记忆T细胞,T cells follicular helper为滤泡辅助性T细胞,T cells regulatory(Tregs)为调节性T细胞,T cells gamma delta为γδT细胞,NK cells resting为未活化的自然杀伤细胞,NK cells activated为活化的自然杀伤细胞,Monocytes为单核细胞,Macrophages M0为M0型巨噬细胞,Macrophages M1为M1型巨噬细胞,Macrophages M2为M2型巨噬细胞,Dendritic cells resting为未活化的树突状细胞,Dendritic cells activated为活化的树突状细胞,Mast cells resting为未活化的肥大细胞,Mast cells activated为活化的肥大细胞,Eosinophils为嗜酸粒细胞,Neutrophils为中性粒细胞;Fraction为分数;红色小提琴代表银屑病皮损,蓝色小提琴代表非皮损皮肤
进一步将上述得到的25个DEGs与22种免疫细浸润胞的特征基因表达量进行相关分析,结果显示 CMAHP、LOC100506990、MTURN、PAPLN、PTPN21、RGMB、RORα、SIK1、WNT2B和ZNF273的表达量均与活化的CD4记忆T细胞、活化的NK细胞和活化的肥大细胞浸润呈显著负相关(P<0.05),见图8。
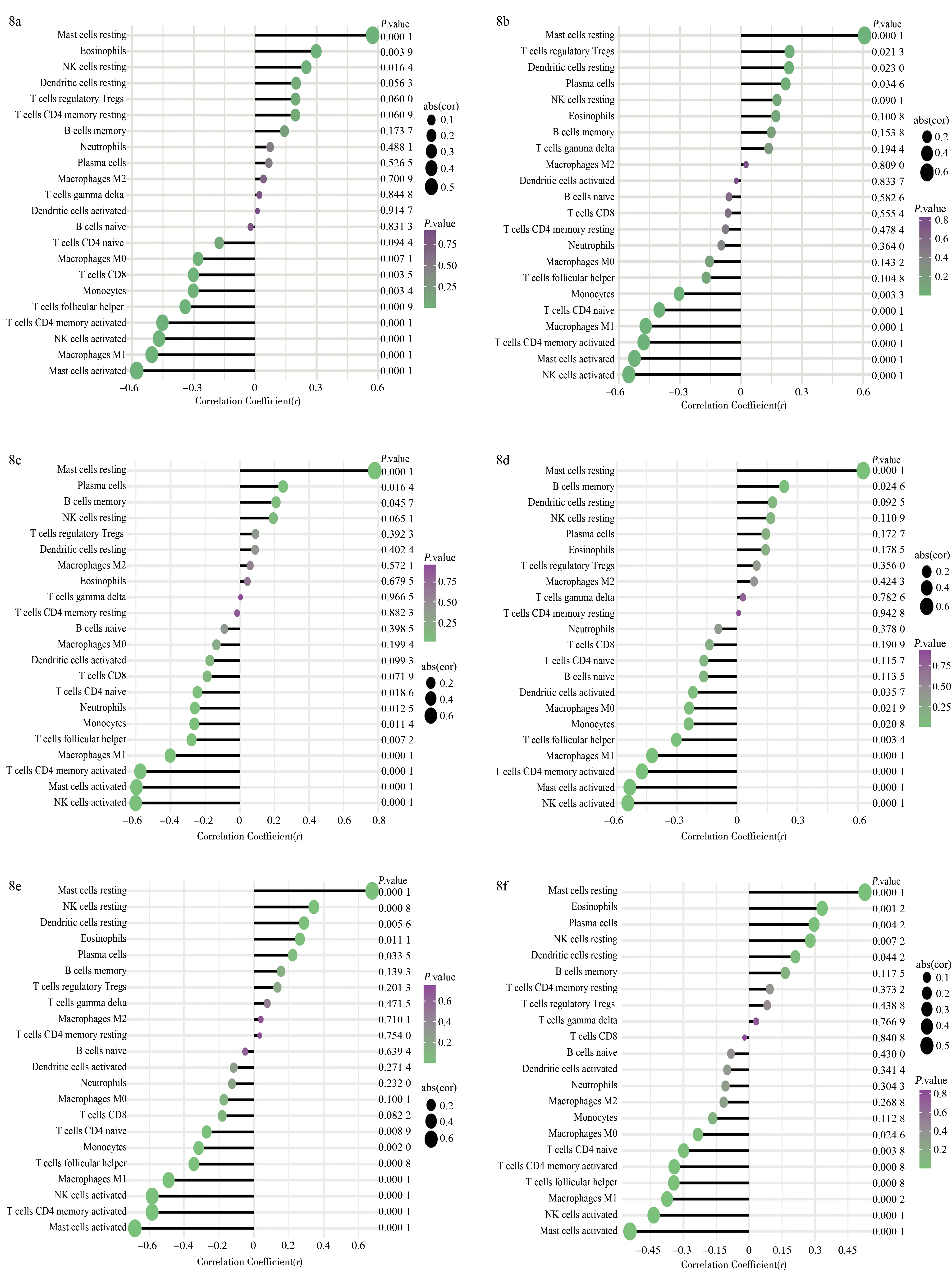
注:Mast cells resting为未活化的肥大细胞,Eosinophils为嗜酸粒细胞,NK cells resting为未活化的自然杀伤细胞,Dendritic cells resting为未活化的树突状细胞,T cells regulatory Tregs为调节性T细胞,T cells CD4 memory resting为未活化的CD4记忆T细胞,B cells memory为记忆B细胞,Neutrophils为中性粒细胞,Plasma cells为浆细胞,Macrophages M2为M2型巨噬细胞,T cells gamma delta为γδT细胞,Dendritic cells activated为活化的树突状细胞,B cells naive为幼稚B细胞,T cells CD4 naive为幼稚CD4 T细胞,Macrophages M0为M0型巨噬细胞,T cells CD8为CD8 T细胞,Monocytes为单核细胞,T cells follicular helper为滤泡辅助性T细胞,T cells CD4 memory activated 为活化的CD4记忆T细胞,NK cells activated为活化的自然杀伤细胞,Macrophages M1为M1型巨噬细胞,Mast cells activated为活化的肥大细胞;CMAHP为单磷酸胞嘧啶-N-乙酰神经氨酸羟化酶假定蛋白,MTURN为神经前体分化调节同系物,PAPLN为原肠(胚)形成糖蛋白,PTPN21为蛋白酪氨酸磷酸酶非受体21,RGMB为排斥性导向分子B,RORα为维甲酸相关孤儿受体α,SIK1为盐诱导激酶1,WNT2B为Wnt家族成员2B,ZNF273为锌指蛋白273;Correlation Coefficient(r)为相关系数,abs(cor)为相关系数的绝对值;点的大小代表基因与免疫细胞之间相关性的强度
3 讨 论
银屑病是一种复杂的多因素疾病,皮肤表现多种多样,自身免疫和自体炎症反应共存[7]。近年来出现了各种治疗银屑病的新方法,但目前银屑病仍不能治愈[8-9]。本研究通过R语言联合WGCNA的方法筛选出89个交集DEGs。KEGG注释显示这些DEGs富集到了IL-17信号通路这条关键的致病通路。研究证明,IL-23/辅助性T细胞(helper T cell,Th细胞)17通路是调控银屑病发病机制中的一个关键途径。抗原呈递、核因子κB信号通路激活、Th细胞分化和IL-17反应增强共同参与银屑病的致病过程,诱导机体免疫反应的发生和免疫细胞浸润[10]。中性粒细胞胞外陷阱的过度表达会刺激IL-17的释放以及炎症介质的合成,进而导致中性粒细胞的自我放大[11]。此外,角质形成细胞产生炎症细胞因子和表达IL-17受体,启动和促进银屑病的发生[12]。在IL-17和炎症因子共同作用下,内皮细胞黏附分子的表达增加[13]。研究证明,肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)抑制剂对IL-17驱动的银屑病的疗效显著[14]。因此,针对TNF-α、IL-23和IL-17的生物制剂已经被开发并被批准用于银屑病的治疗。
免疫反应紊乱是银屑病炎症发生和维持的原因,而持续炎症通过TNF-α、IL-17和γ干扰素导致角质形成细胞不受控制的增殖和分化功能障碍[15]。值得关注的是,本研究GSEA交集通路的结果显示,筛选出的DEGs均抑制了嘧啶代谢、利什曼原虫感染、亚油酸代谢、细胞因子受体相互作用、趋化因子信号通路和嘌呤代谢上述信号通路,说明这些DEGs可能对银屑病的发生发展起重要的抑制作用。
T细胞信号在银屑病的发病机制、治疗和共病方面发挥至关重要的作用。先天免疫系统中多种免疫细胞在银屑病皮损中显著增加,如角质形成细胞、树突状细胞、T淋巴细胞、中性粒细胞、巨噬细胞等贯穿寻常型银屑病的发病过程。皮损部位角质形成细胞可通过释放趋化因子招募T细胞聚集[16],产生IL-17A和IL-23,并与产生CD8+T淋巴细胞的获得性免疫反应细胞(如Th17细胞和IL-17)相互作用[17]。银屑病的免疫学紊乱与T细胞亚群以及相应的细胞因子(如γ干扰素、TNF-α、IL-23和IL-17)的过度刺激有关[18]。因此,为了进一步探讨免疫细胞浸润在银屑病中的作用,本研究对DEGs与22种免疫细浸润胞的特征基因表达量进行相关分析,结果显示,初始B细胞、记忆B细胞、浆细胞、CD8 T细胞、初始CD4 T细胞、未活化的CD4记忆T细胞、活化的CD4记忆T细胞、滤泡辅助性T细胞、γδ T细胞、活化的NK细胞、单核细胞、M0型巨噬细胞、M2型巨噬细胞、未活化的树突状细胞、活化的树突状细胞、活化的肥大细胞、嗜酸粒细胞和中性粒细胞的浸润增加,而调节性T细胞、未活化的NK细胞、M1型巨噬细胞和未活化的肥大细胞的浸润减少可能与银屑病的发生发展有关。这些关联的具体机制还需要进一步的实验证据进行验证。本研究显示,CMAHP、LOC100506990、MTURN、PAPLN、PTPN21、RGMB、RORα、SIK1、WNT2B和ZNF273对抑制免疫细胞浸润发挥重要作用。
GO注释显示RORα均较好地关联到与银屑病发病紧密相关的生物学功能上。在免疫系统中,RORα作为炎症关键负调节因子,在淋巴细胞和骨髓细胞均有表达,其与IL-17A、IL-23R的表达和Th-17细胞的分化有关[19-20]。SIK1负调节Toll样受体4诱导的核因子κB的激活,减少促炎性细胞因子的表达,其抑制作用可将巨噬细胞再转化为抗炎表型[21-22]。研究发现,PTPN21可能与肿瘤的发生和预后有关,其过表达可激活促分裂原活化的蛋白激酶信号通路,促进细胞增殖[23-24]。骨髓间充质干细胞过度表达PTPN21后,免疫抑制功能受损,肿瘤细胞和血管内皮细胞的募集能力增强[25]。有研究显示,MTURN在肺癌组织中表达上调,可作为肺癌的潜在生物标志物[26]。此外,PAPLN过表达则提示急性髓系白血病髓外浸润的预后较好[27]。WNT2B在宫颈癌细胞中表达水平上调,逆转了某些微RNA对癌细胞增殖和转移的抑制作用以及并促进了相关细胞的凋亡[28]。
综上所述,本研究采用GSEA联合WGCNA和CIBERSORT算法等生物信息学分析手段,筛选和挖掘了与银屑病发病相关的DEGs(CMAHP、LOC100506990、MTURN、PAPLN、PTPN21、RGMB、RORα、SIK1、WNT2B、ZNF273),这些靶基因可能在银屑病的发病过程中起保护作用,这将有助于后续更加深入地探讨其致病机制,为银屑病的临床诊断、治疗提供新思路。
