一种高通量自动化血浆miRNA 文库构建方法及其跨批次性能评估
2022-06-23么欣彤孙善月刘雅晴石乐明郑媛婷
么欣彤 孙善月 刘雅晴 石乐明 郑媛婷
(复旦大学生命科学学院人类遗传学与人类学系 上海 200438)
生物标志物的发现为疾病早期诊断、预后评估、治疗方案选择以及疗效监控[1-3]带来新的希望,是实现精准医学的基石[4]。micro RNA(miRNA)是一种长度为21~23 个核苷酸的非编码小RNA 分子,通过与Argonaute(AGO)蛋白形成miRNA 介导的沉默复合物(miRNA induced silencing complex,mISC)抑制基因表达[5]。血浆来源的miRNA 的表达水平与疾病生理状态息息相关[5-9],并具有易采样、稳定性高、可快速定量等优点,已成为临床生物标志物的重要候选来源。例如,肝癌分子诊断试剂盒miRNA7™使用7 种血浆miRNA 作为标志物,实现了肝癌的精确诊断[10]。
miRNA 测 序 技 术(microRNA sequencing,miRNA-seq)是一种基于高通量测序技术的miRNA序列分析方法,由于准确度高、检测范围广等优势已成为发现miRNA 生物标志物的重要手段[11-12]。立足于大规模临床队列的血浆样本,研究者可使用miRNA-seq 对患者组和对照组的血浆样本中miRNA 进行全面的定量,寻找组间差异miRNA 作为潜在的生物标志物。因此,高质量的miRNA-seq数据是生物标志物挖掘的必要前提[13-14]。miRNAseq 质量控制研究基于人工合成的小RNA 序列或单个生物样本对不同建库方法的检出灵敏度、定量准确性与可重复性进行评估[13,15]。然而,目前尚缺乏对于miRNA-seq 技术批次内性能的客观评估及跨批次稳定性的质量控制研究。与RNA-seq 相似[16],miRNA-seq 数据也面临着批次间存在系统性差异的问题[11],即批次效应。而生物标志物的研究和验证往往依赖于超大型队列[5],样本的文库构建需要分为多批次进行,严重的批次效应可能导致生物信号被覆盖,因此跨批次定量稳定性对于miRNA-seq 定量方法是十分必要的。但是miRNAseq 文库构建实验流程复杂、文库质量极易受到操作细节的影响,传统的人工操作由于通量低、易出现操作错误等限制,难以满足大型队列文库构建的需求。因此,亟需一种自动化建库方法实现高效、跨批次稳定的文库构建实验。
本研究基于自动化工作站建立一种高通量、自动化的血浆文库构建方法,并使用自制的3 种血浆参考物质,基于该自动化建库方法分4 批次产生了32 个miRNAs-seq 文 库,从miRNA 检 出 种 类、绝 对定量、相对定量、差异表达分析等层面评估方法性能及跨批次稳定性。
资料和方法
Sciclone®NGSx 自动化液体工作站Sciclone®NGSx 自动工作站(美国PerkinElmer 公司)是一台可以实现温度控制与精确移液的液体工作站。该工作站由样品操作台、机械工作臂、温度控制等功能模块及一台计算机构成,可通过编写程序控制工作站运行,模拟人工操作流程进行文库构建。该工作站每批次可构建96 个文库。除实验过程中涉及的试剂配制、部分样品孵育环节需要在PCR 仪上进行以外,其余步骤均可在Sciclone®中自动完成。
血浆参考物质本研究制备了3 类血浆参考物质,分别命名为P10、P11 和PM。其中P10、P11 分别为1 名健康男性志愿者和1 名健康女性志愿者血浆,均为本课题组成员;PM 为2 型糖尿病患者的血浆样品混合物,来自于上海市市级重大专项 “国际人类表型组计划(一期)” 项目(Grant No.2017 SHZDZX01)——伴随西格列他钠Ⅲ期临床试验的多组学研究[17]。本研究经复旦大学生命科学学院伦理委员会批准(批件号:BE2050),所有研究对象均志愿参加并签署知情同意书。血浆制备方法为:使用真空采血管(EDTA-K2 抗凝)采集2 名志愿者静脉血约50 mL。4 ℃下2 000×g离心10 min,收集上清液至50 mL 离心管;上清液在4 ℃下3 000×g离心10 min,收集上清液至一个新的50 mL 离心管中,获得血浆。为避免样本反复冻融,将血浆分装至1.5 mL EP 管中(200 μL/管)。液氮速冻,于-80 ℃保存。
血浆miRNA 抽提取200 μL 血浆样本于4℃下解冻。使用QIAcube 全自动核酸纯化系统(凯杰生物科技有限公司)配合miRNeasy Serum/Plasma Advanced Kit 试剂盒(凯杰生物科技有限公司),根据标准实验流程完成miRNA 抽提。25 μL 无酶水洗脱,获得约22 μL 血浆小RNA 溶液。分装至2 个1.5 mL EP 管中(11 μL/管),于-80 ℃保存。
自动化miRNA 文库构建使用铂金埃尔默股份有限公司(PerkinElmer®)的NEXTFLEX®small RNA-seq 试剂盒配合Sciclone®NGSx 自动工作站,以10.5 μL 抽提产物为起始物质,使用优化后小RNA 文库构建程序完成文库构建实验。该实验主要分为7 个环节:3′端接头连接(3′-adapter ligation)、未连接的3′端接头去除(excess 3′-adapter removel)、未连接的3′端接头失活(excess 3′-adapter inactivation)、5′端接头连接(5′-adapter ligation)、逆转录(reverse transcription)、磁珠纯化(beads cleanup)、文库扩增(library amplification)和扩增后磁珠纯化(beads cleanup)。除以下操作外,均按照说明书操作:在3′端接头连接和5′端接头连接环节中,将3′端接头与5′端接头4 倍稀释后加入到反应体系中;在文库扩增环节进行25 轮PCR 循环;在扩增前后磁珠纯化的环节中,取消片段筛选过程,使用磁珠法对逆转录或文库扩增产物中所有cDNA 分子进行全回收。最终共得到12 μL 文库产物,装入0.5 mL EP 管中,于-20 ℃保存。
miRNA 文库质检与测序使用赛默飞世尔科技(中 国)有 限 公 司 的Qubit 荧 光 计(Qubit®3.0 Fluorometer)配 合Qubit®dsDNA HS Assay Kit 试剂盒测量文库浓度,并计算文库产量。文库测序由明码(上海)生物科技有限公司完成,在Illumina Hiseq 平台上进行双端150 bp 测序。测序结果为每条读段的原始序列,以fastq 格式存储。
序列比对、计数与标准化本研究使用exceRpt数据预处理流程[18]对每个文库的测序数据进行序列比对与计数;该流程可根据给定序列切除3’端接头序列,过滤掉低质量的读段,去除样本中比对到核糖体RNA 及外源污染物RNA(NCBI UniVec)的序列。然后将读段比对到人类参考基因组(Human reference genome build Genome Reference Consortium GRCh38,UCSC hg38)和miRBase version 21,统计每个测序文件中的miRNA 读段数目。
在原始读段计数基础上,本研究使用CPM(Count Per Million)法对每个文库的原始表达量进行标准化:CPM=(计数+1)/(总miRNA 读段数),并进行log2转化。得到用于后续分析的log2CPM表达谱。
检出可重复性计算方法使用并交比(Jaccard Index)衡量两样本之间(差异检出)miRNA 检出一致性。并交比是指两个样本miRNA 检出集合的交集与两样本miRNA 检出集合的并集大小之比,公式如下:

其中A 和B 分别代表两样本miRNA 检出集合。并交比指数的取值范围是0~1,越接近于1,说明两样本miRNA 检出一致性越高;反之,越接近于0,则说明检出一致性越低。
结 果
血浆miRNA 自动化文库构建方法的建立及其性能为了产生高质量的血浆miRNA-seq 测序数据,便于评估跨批次稳定性,本研究对厂商提供的原始商业化自动化建库方法进行了优化。
首先,为了评估自动化建库工作站产生的miRNA-seq 文库质量,本研究使用人工操作和自动化方法对3 类血浆参考物质(P10、P11 和PM)纯化得到的小RNA 进行平行建库,共产生87 个miRNAseq 文库。我们将自动化工作站操作产生的74 个文库和人工操作产生的13 个文库分别称为自动文库和手动文库,并从文库产量(图1)和对不同生物样本区分程度(图2)对两类文库进行质量评价。一方面,自动文库的产量极低[(8.5±5.6)ng],仅为手动建库文库产量[(105.6±66.2)ng]的8%。另一方面,对13 个手动文库进行主成分分析,可以观察到来自相同血浆样本的文库聚集在一起,而来自不同样本的文库明显分开(图2A)。然而,自动文库却不能区分来自相同或不同血浆样本的文库(图2B),这意味着使用自动化工作站得到的文库测序数据噪音过大,甚至超过了不同样本之间的固有生物学差异。以上证据共同表明,厂商提供的原始商业化自动化建库程序不能很好地模拟miRNA-seq 手动建库,所得到的miRNA 文库质量低下,无法满足需求。

图1 优化前自动化文库与手动文库文库产量对比图Fig 1 Comparsion of yields between libraries constructed with manual protocol and vendor-provided automated protocols
为解决这一问题,我们对影响文库质量的关键因素进行了优化(表1),并使用优化后的自动化建库方法构建了4 个批次(编号为a、b、c、d)的血浆参考物质miRNA 文库,每批次8 个样本(3 个P10、3 个P11 和2 个PM 样本)。与原始自动化程序(vendorprovided automated protocol)相比,优化后的自动化程序(optimized automated protocol)文库产量提升近4 倍(表2);主成分分析和无监督聚类结果显示,优化后来自相同血浆样本的不同批次的技术重复优先聚在一起(图2C、图3),而来自不同血浆样本的文库清晰地分开,说明优化后自动化方法对不同生物样本的miRNA 表达差异具有良好的区分能力,可用于跨批次稳定性质量评估。

图3 采用优化后自动化建库方法在不同批次分析血浆参考物质的miRNA 表达矩阵热图Fig 3 Heatmap of miRNA expression matrix of plasma reference materials profiled in multiple batches with the optimized automated library construction protocol
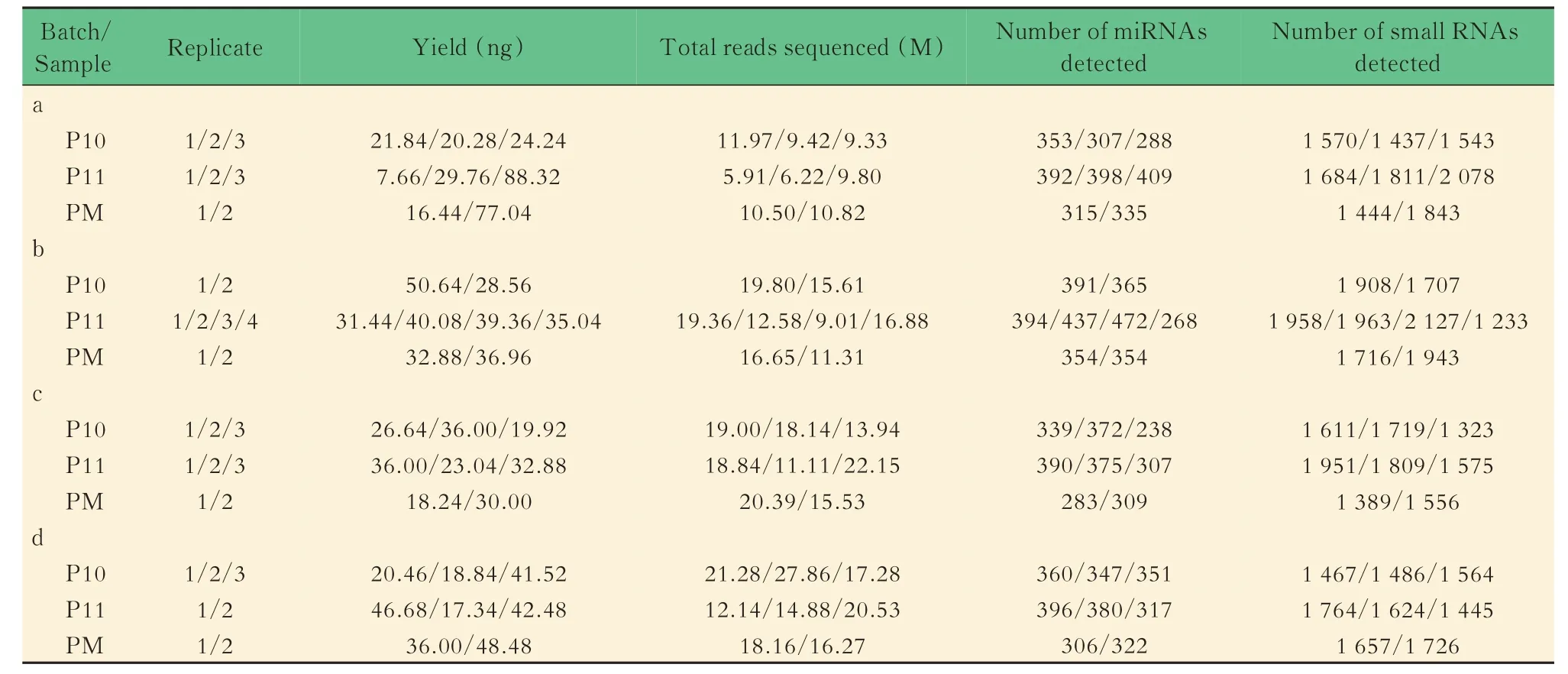
表2 优化后自动化文库质量基本情况Tab 2 Quality overview of the libraries constructed with optimized automated protocol

图2 优化后自动化建库方法对不同生物样本具有更好的区分能力Fig 2 Optimized automated library construction method demonstrated improved power in discriminating biologically distinct groups of samples

表1 自动化建库程序优化参数总结表Tab 1 Parameter optimizations and their impacts on library quality
评估自动化方法miRNA 检出的跨批次稳定性同类样本miRNA 检出的稳定性是定量结果可靠的前提。因此,我们从miRNA 检出数目和检出种类两个层面对miRNA检出的跨批次稳定性进行评估(图4A)。
3类血浆样本miRNA检出种类基本情况如(图4B)所示。不同批次的文库中检出的miRNA 数目基本稳定,约 为300~400 种。P10、P11、PM 样 本 检 出 的miRNA 种类分别为337±44、380±54 和322± 25。
为评估miRNA 检出种类在批次间的一致性,我们使用并交比(Jaccard Index)定量描述两次实验中检出的miRNA 种类的相似度。通过计算所有样本任意两两配对(C232次比较)检出种类的并交比,对跨批次的技术重复之间的并交比与同批次的不同技术重复之间的并交比进行比较(图4A)。首先,同类血浆样本的不同技术重复之间的检出一致性显著高于不同血浆样本之间检出一致性(图4C),说明不同血浆样本中表达的miRNA 种类具有一定差异;对于同种血浆参考样本而言,跨批次技术重复之间的并交比与同批次内技术重复之间并交比无统计学差异(图4D),表明本研究中产生的4 批文库在miRNA 检出层面无明显的批次效应。

图4 评估miRNA 检出的跨批次稳定性Fig 4 Cross-batch concordance of miRNA detection
评估miRNA 绝对定量的跨批次稳定性技术重复间绝对表达水平的一致性是定量可靠性的前提。为评估miRNA 绝对定量的跨批次稳定性,我们使用两样本miRNA 表达向量的皮尔森相关系数(Pearson correlation coefficient)作为衡量两个样本之间miRNA 检出绝对定量一致性的指标,对于每种血浆样本,我们对跨批次样本间与同批次样本间的绝对定量相关系数进行t检验(图5A)。P10(P=0.26)和P11(P=0.24)样本的批次间一致性与批次内一致性差异无统计学意义,而对于PM(P=0.02),批次内相关系数略高于批次间,可能与PM 样本技术重复数目少有关。同一批次不同技术重复之间的一致性与不同批次技术重复之间的一致性差异无统计学意义(P=0.80),我们计算得到所有样本两两间相关系数矩阵,并基于该相关系数矩阵进行无监督层次聚类。结果显示:来自相同血浆的样本优先聚类(图5B),而来自相同批次的样本无明显聚集。这表明批次定量差异具有随机性,且小于不同生物学样本间的固有差异。本研究中产生的4 批文库在绝对定量层面具有良好的跨批次稳定性。

图5 评估miRNA 绝对定量的跨批次一致性Fig 5 Cross-batch concordance of absolute-expression measurements
评估两样本间相对定量的跨批次稳定性绝对定量一致性仅能表明一种方法对单类生物样本具有良好的测量可重复性,而生物标志物的筛选往往依赖于稳定、可靠地检测出不同生物样本组间miRNA 表达水平差异。因此,我们对两组生物样本间miRNA 相对定量的可重复性进行评估。
使用limma 软件包分别对3 个批次文库的P10样本与P11 样本(P10/P11)进行差异表达分析:每个批次内P10 样本和P11 样本的3 个技术重复进行差异表达分析,共进行3 次差异表达分析,得到的log2FC 向量表示该批次两样本间的相对表达水平。我们仅使用了a、c 和d 3 个批次的文库,批次b 只有2 个P10 样本而没有被纳入)。我们使用两个批次log2FC 向量之间的皮尔森相关系数衡量批次间相对定一致性。3 个批次两两之间的相关系数的分别为0.69、0.75 和0.79(图6),表明P10/P11 样本的批次间相对定量一致性较好。
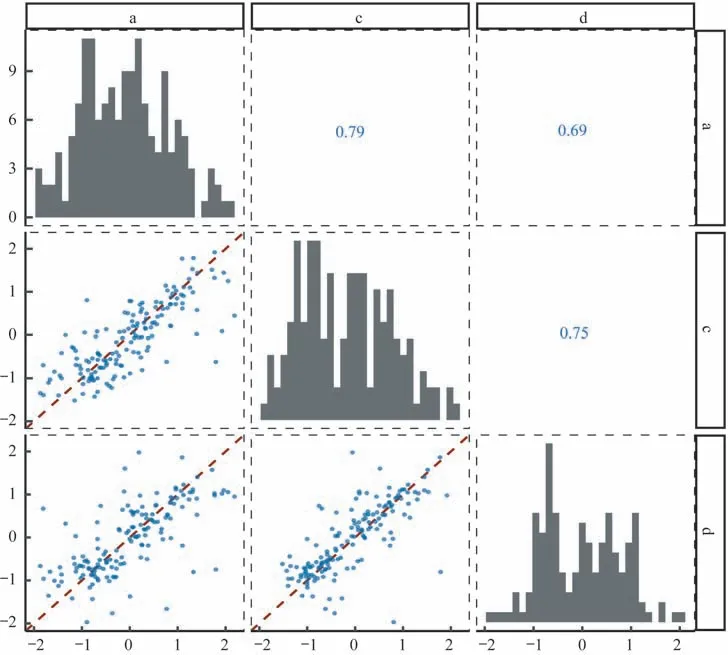
图6 跨批次P10/P11 相对定量一致性Fig 6 Cross-batch concordance of relative-expression between P10 and P11 samples
评估两样本差异表达分析的跨批次稳定性为评估检测组间差异表达miRNA 的可靠性,我们评估了批次间差异表达结果的一致性。本研究使用limma 包分别鉴定了每个批次P10 样本和P11 样本之间的差异表达miRNA[P<0.05,且|log2FC|>log2(1.5)]。
每个批次分别检测到63、68 和61 个差异表达miRNA(图7A),共检测到44 个上调miRNA,其中27 个(61.3%)可以在至少两个批次中检出;共检出49 个下调miRNA,其中33 个(67.3%)可以在至少两个批次中被检出(图7B),说明生物样本之间跨批次差异表达分析结果具有较好的一致性。
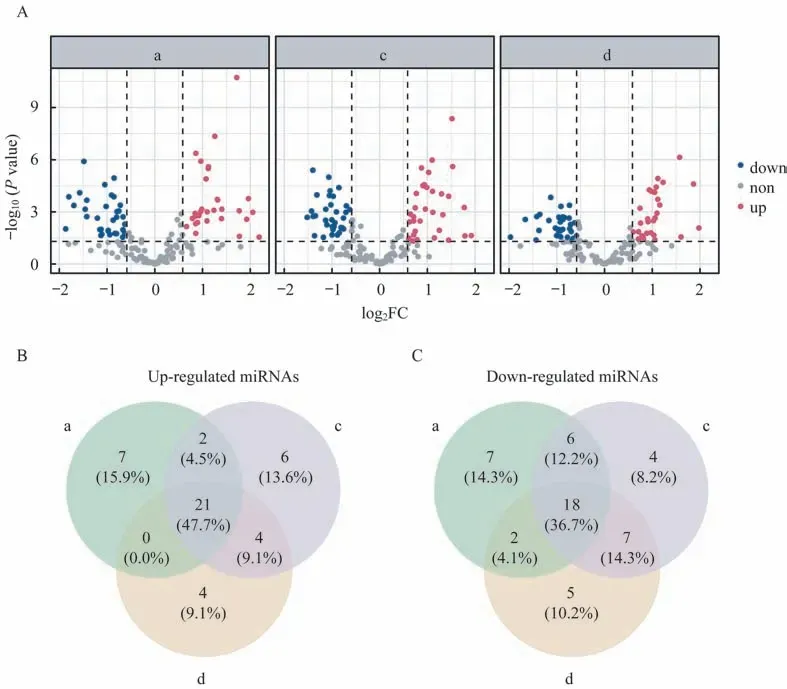
图7 跨批次P10/P11 差异表达miRNA 一致性评估Fig 7 Cross-batch concordance of differentially expressed miRNAs between P10 and P11 samples
讨 论
miRNA-seq 技术可以不基于任何先验知识而描绘样本中miRNA 组学全貌,且测序成本不断降低,从而为miRNA 生物标志物的发现提供有力的工具。产生上千例样本的大型队列miRNA-seq 数据需要高效、性能可靠的文库构建方法。目前已有多种自动化液体工作站,可适配小RNA 文库构建试剂盒、实现自动化文库构建。但是,自动化建库系统受到操作精细程度与灵活程度的限制,难以模拟人工建库操作流程,而自动化平台所构建的文库质量是否与人工建库可比又是自动化建库方法必须回答的问题。综上,在将自动化文库构建方法应用于大规模临床样本测试之前,需要建立系统的质量评估方法对其进行性能验证。
跨批次性能评估方法的建立目前对miRNA定量方法的质量评估主要基于合成RNA 片段与单个生物样本的miRNA 定量可重复性、准确性、敏感性 和特异性[15,19]。miRQC 研 究[20]与SEQC 研究[13]使用两类生物样本参考物质(sample A 和sample B),不仅评估了绝对定量的准确性和可重复性,而且对样本间相对定量、差异表达的可靠性进行评估,表明:(1)检测不同生物样本间差异的能力是评估一种定量方法的重要指标[13,19];(2)使用不同类型的参考样本或从不同的评估角度出发,方法评估的结果与结论可能完全不同,因此需要基于与待测样本种类相同的参考样本,建立适合的质量评估方法[13,20]。
本研究建立了一套基于3 类真实的血浆参考物质的性能评估方法,以参考物质之间miRNA 表达水平的相对差异作为主要质控指标来评估定量方法的性能。3 类样本分别来自于一名健康男性、一名健康女性志愿者和糖尿病患者混合血浆,以模拟临床队列中不同性别和健康状态的群体。基于该套参考样本集,不仅可以考察单一样本绝对定量可重复性、两样本之间相对定量和差异表达的可靠性,还可以考察定量方法对不同种类生物学差异的区分能力。图2C 主成分分析结果显示,PC1 可以区分不同性别的样本,PC2 可以区分糖尿病和健康人的样本,提示优化后自动化建库方法可能对miRNA表达的性别差异及疾病状态差异具有良好的区分度。该思路对于miRNA 质控领域具有参考价值。
此外,当前miRNA 质控研究主要关注跨平台、跨实验室的性能比较[15,19],对跨批次稳定性却鲜有研究。本研究使用相同的参考样本集连续进行4 个批次的文库构建实验,每个批次内同类血浆样本设计2~3 个技术重复。通过设计批次内和批次间技术重复,并以批次内技术重复一致性为基准,从而客观、有效地评估该定量方法的跨批次一致性。本研究提供了一种跨批次稳定性的评估方法,对于大型队列数据产生平台的质量评估具有参考价值。感兴趣的读者可以从作者处免费获取基于三类参考物质的miRNA 表达谱数据。
血浆miRNA 自动化文库构建方法的建立本研究通过上述基于3 类真实血浆参考物质的跨批次一致性评估方法,以参考物质之间miRNA 表达水平的相对差异作为主要质控指标和优化目标,有效地发现出商业产品中所存在的且之前不为厂商所认识到的严重问题:在200 μL 血浆miRNA 文库构建实验中,使用厂商提供的自动化程序建库与人工操作的文库质量具有很大的差距(仅为手动建库文库产量的8%);更为严重的是,由此导致该方法不能全面地刻画出每种生物学样本独特的miRNA 表达特征,因而无法捕捉到不同类样本间内在的生物学差异。因此需要确定厂商提供的自动化流程中导致文库质量降低的关键因素并加以修正。
为此,本研究设计了一系列探索试验,发现并确证了自动化建库流程中影响文库质量的关键因素,主要涉及3 个方面:(1)在磁珠纯化环节中,取消去除长片段的磁珠筛选步骤,可以使得文库产量提高1.9 倍(表1)。这意味着对于缺乏长片段核酸分子的血浆样本而言,多余的磁珠筛选环节不但不能通过弃掉长片段富集目标产物(插入miRNA 序列的短片段),反而引起目标产物(短片段)的丢失。然而,对于细胞样本等富含长片段分子的样本而言,磁珠筛选环节对于富集目标产物又是非常必要的。因此针对特定样本的特征动态调整实验方案非常必要;(2)在未连接的3’端接头去除环节,通过调整ADS、Isopropanol 试剂体积及Elution buffer 种类,使之与人工建库实验条件保持一致,可以使得文库产量提升5 倍(表1)。表明在自动化方法的开发过程中,对于任何操作细节的变动都需要非常谨慎,一些细微的差异即有可能引起产物的随机丢失,导致自动化文库质量大大降低,无法捕捉到样本miRNA 转录组中的全面信息;(3)在未连接的3’端接头失活、5’接头连接及逆转录环节,将原自动化流程中的孵育条件(在工作站内温控模块孵育)与人工实验中的孵育条件(在PCR 仪中孵育)保持一致,可以使得文库产量提高1.4 倍,RNA 检出种类提高1.1 倍(表1)。表明建库实验中孵育过程的温度控制对于反应效率非常重要,在仪器开发过程中需要特别关注温控装置的性能验证。本研究揭示了自动化建库中影响文库质量的关键因素,对于自动化建库方法的开发具有借鉴意义。尽管优化后方法信噪比可以达到手动文库的水平,但自动建库文库产量与手动文库仍有约3 倍差距,说明该自动化操作方法的细节还有优化的空间。
综上所述,本研究以重要实际需求为导向,针对商业化的自动化血浆miRNA-seq 文库构建过程中所存在的严重质量问题而展开。通过采用多类血浆参考物质,并以参考物质之间miRNA 表达水平的相对差异作为主要质控指标和优化目标,发现出商业产品中所存在的严重问题。为解决该问题,我们建立了一种高通量、自动化miRNA 文库构建方法,可跨批次稳定地产生大批量血浆样本的miRNA 数据,适用于大型临床队列血浆miRNA 文库构建试验。目前复旦大学生科院人类表型组研究院已将本方法应用于国际人类表型组计划,产生了多个队列共5 000 多个血浆样本的miRNA 表达谱数据,为生物学和基础医学研究提供了有力的技术支持。
作者贡献声明么欣彤 论文构思、撰写和修订,数据产生和分析,绘制图表。孙善月 数据产生,分析结果确认。刘雅晴 论文修订,分析结果确认。石乐明,郑媛婷 课题构思和修订。
利益冲突声明所有作者均声明不存在利益冲突。
