活化H2O2的芬顿/类芬顿体系的研究现状
2022-06-22俞乙平林少华钱子姣
俞乙平,林少华,钱子姣
(南京林业大学 土木工程学院,江苏 南京 210037)
随意排放的污废水会导致严重的水污染。传统的废水处理方法通常是先经过生物降解法和/或物理化学法(如絮凝、氯氧化和臭氧氧化等),再进行过滤和吸附分离等处理,但这些传统工艺方法只能处理一些简单废水,不适用于处理较复杂的含有毒有害有机物的废水。
相比之下,高级氧化技术因其普遍有着高稳定性、高降解率、无二次污染等优点而越发受到人们的青睐[1]。它可在接近环境温度和压力下工作,产生的强氧化自由基(主要是羟基自由基HO·)能将有机污染物完全分解为CO2、H2O等无毒物质。在过去二十年中,各种基于HO·生成的高级氧化技术已经被广泛发掘,并根据自由基的主要来源进行分类,分为催化湿式氧化法、臭氧氧化法、电化学氧化法、芬顿/类芬顿法、光化学氧化法等。其中芬顿/类芬顿法是研究较早且应用比较广泛的高级氧化法之一,因其在废水处理上的降解效果良好,受到了人们的广泛青睐。
本文阐述了芬顿技术的发展及现存问题,介绍了针对这些问题所改进的类芬顿技术。但是铁基催化剂有许多问题还是不可避免的,所以本文进一步重点介绍了非铁基芬顿催化剂在应用时的氧化还原特性、优势和潜在应用领域。最后,分析了各非铁基催化剂在实际应用时的不足,并对其未来技术发展做出了展望。
1 经典和改进的类芬顿反应
1.1 经典芬顿
经典的芬顿反应是通过亚铁离子(Fe2+)激活过氧化氢(H2O2)并通过一系列反应产生出羟基自由基(HO·)。
但经典芬顿法存在以下许多缺陷:(1)H2O2的储存和运输存在较高的成本和风险;(2)使用的铁离子容易形成铁污泥,需要及时处理掉;(3)反应在酸性pH下操作,但酸化反应后,需要中和;(4)对反应设备要求高,限制了其在实际工程中的应用。
1.2 改良的类芬顿法
为了提高氧化效率,已经在经典芬顿反应上做了许多改进,统称为“类芬顿”反应[2],即通过 H2O2产生·OH而促进有机物分解的方法。类芬顿反应克服了传统芬顿反应的部分缺点,具有pH值范围广、铁污泥减少、微量高效等优点,具体可以分为光芬顿、电芬顿和超声波-芬顿法。
1.2.1 光芬顿 在“光芬顿”过程中,在λ<260 nm下,使用紫外线(UV)或太阳光照射Fe2+和H2O2溶液,可以加速Fe3+和Fe2+之间的相互转化,Fe3+的光还原与H2O2光辐射分解的协同作用获得大量的·OH。光芬顿反应不仅减少了铁污泥的生成,还可更高效的降解废水中的污染物。
在染色有机废水中,会发生染料敏化光引发的光芬顿反应。通过可见光照射可以使分子间电子转移增强,从而将Fe3+还原为Fe2+,并显著提高HO·生成速率。
但是,光芬顿反应存在光能利用率低、消耗能量大、成本较高的缺点[3]。因此,提高光芬顿反应中紫外线的利用率以及充分利用好太阳能的光源是推动光芬顿广泛应用的重点。
1.2.2 电芬顿 为了克服这些缺点并提高污染物去除效率,学者们研究出了基于芬顿反应的电化学光催化剂,即“电芬顿”反应。它通过供给纯氧或者空气在阴极上原位并连续电生成H2O2,再向溶液里加入Fe2+催化剂,并将Fe3+阴极还原成Fe2+,随后连续生成芬顿试剂。
电芬顿因其处理效率高、无二次污染、便于操作等优点被人们熟知,多被于废水处理上。但是电芬顿仍存在处理效率低等问题,而且需要在酸性pH条件下反应,因此探索性能良好的电极和拓宽pH值具有重要意义。
1.2.3 超声波-芬顿法 在超声波-芬顿法中,水和分子氧的超声波分解不仅增强了HO·的生成,还原位生成了H2O2。
超声波-芬顿法可以提高传质效率,但是超声波的功率消耗较大,且在长时间超声波的高频振动下,对反应器有较大的损耗。所以,探寻能耗较低的超声波仪器和抗震的水处理构筑物尤为重要。
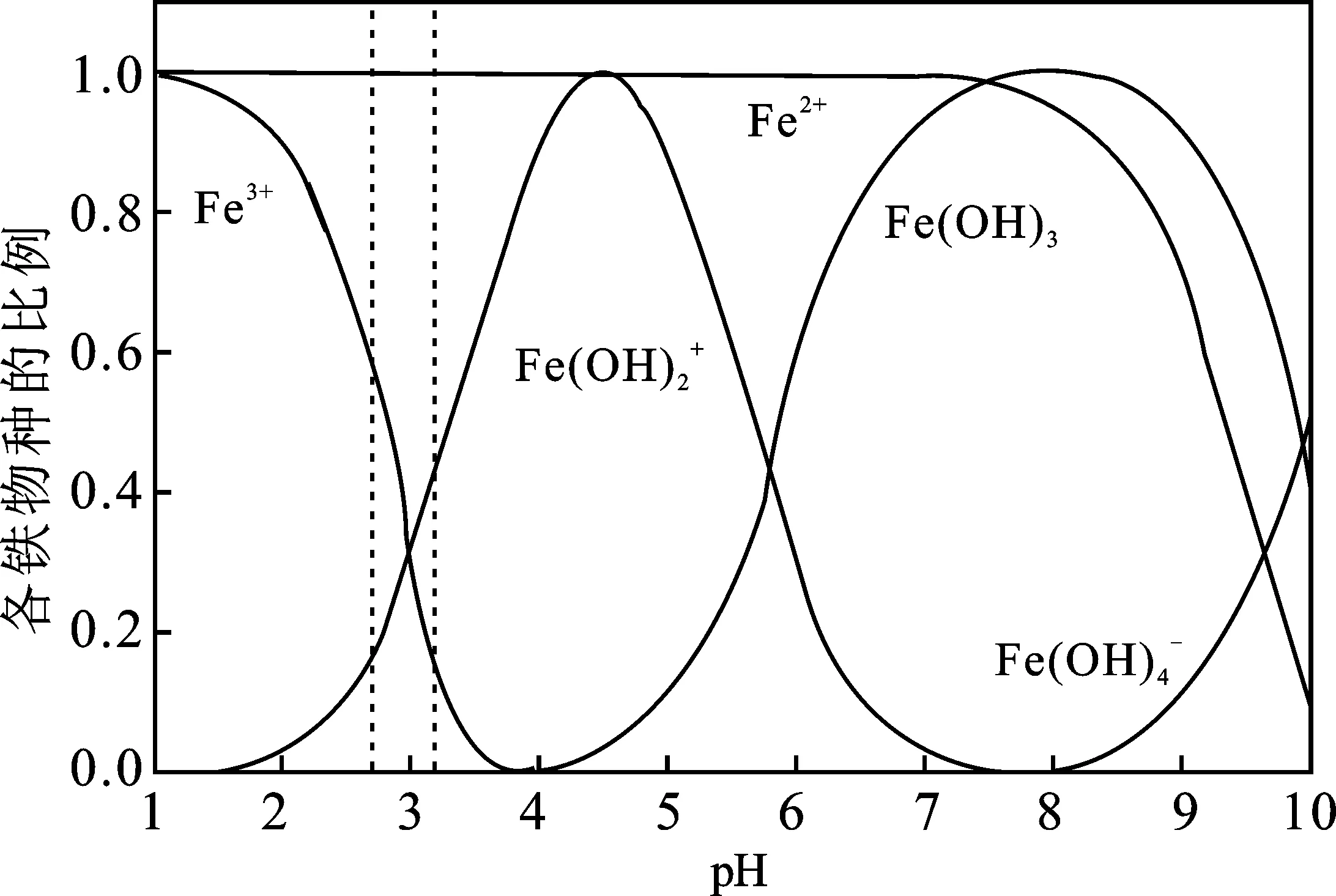
把类芬顿反应组合所形成的混合体系也可称为改性芬顿反应,组合后的芬顿反应所表现的优势更加突出。Francisca-C Moreira等[4]比较了电芬顿(EF)、光电芬顿(PEF)和太阳能下的光电芬顿(SPEF)的降解垃圾渗滤液的能力,发现其降解率和HO·的生成量呈现EF 表1 各种经典芬顿和类芬顿反应的总结Table 1 Summary of various classical and improved Fenton reactions 在高级氧化法中,芬顿技术是应用最广泛的工艺,而铁基催化剂是芬顿技术中应用最频繁的催化剂,因为铁基催化剂有许多优势:(1)高丰度低价格,简单易得;(2)环境相容性高,低毒性;(3)Fe2+和Fe3+易转化。 但是,Fe2+、Fe3+和氢氧化铁的特性都依赖于pH的变化,见图1。Fe2+在中性酸碱度下依旧保持着高溶解度,但Fe3+的溶解度在pH≥4时几乎消失,易形成铁污泥。因此,实际应用中须严格控制低于pH=4的酸性条件下反应,并且需要中和后再排放,所需成本较高[12]。而在紫外线或太阳能照射下,这些氢氧化物的络合物会被回收和再利用,光诱导的芬顿反应减少了铁污泥的生成和Fe2+的输入,提高了有机污染物的降解速率[13]。铁污泥中光惰性络合物的存在、高污泥浓度下光穿透性的降低以及质子对·OH的清除增加等副作用都限制了光芬顿的大规模应用[12]。因此,只有在pH=3时,Fe2+、Fe3+和[FeⅢ(OH)]2+这三种活性物种才能存活,为了保持着pH=3的酸碱度,此时需要加入大量的硫酸,所以这种均相芬顿法是不切实际的。 图1 不同pH值下铁物种的比例[14]Fig.1 Iron species in aqueous solution as a function of pH 为了避免基于均相芬顿试剂的AOPs法受到pH限制,可使用非均相芬顿法中的固体催化剂,这些催化剂主要是稳定的含铁物质(主要是Fe3+)。零价铁[15](ZVI)、黄铁矿(FeS2)、氧基氯化铁(FeOCl)、磁铁矿[16](Fe3O4)、磁赤铁矿[17](γ-Fe2O3)、赤铁矿(α-Fe2O3)和针铁矿[18](α-FeOOH)是研究最广泛的非均相芬顿法的铁基催化剂,沸石、柱撑夹层粘土[19]、nafion膜、活性炭、皂土等[20]也是有效的芬顿催化剂。 然而,在非均相铁基催化剂下,Fe3+和H2O2之间的反应比经典芬顿反应至少慢了3个数量级,可通过紫外线照射加速Fe2+的还原。但低效的光吸收会降低整个过程的效率,在保证H2O2产量的同时还需提高其分解利用率。另一方面,在不调整pH值的情况下,同时,固体催化剂的中金属离子会泄露,容易形成污泥。 由于均相和非均相铁基芬顿法在实际应用中都存在严重的不足,所以需寻找既实用且经济的芬顿的催化剂。为了实现高效的使H2O2分解成·OH,芬顿催化剂应表现出多种氧化态,因为具有特定氧化态的催化活性物种可以通过简单的氧化还原反应循环再生。并且,活性和非活性物种都应该在较宽的pH范围内保持稳定,以防止催化物种的沉淀。 非铁类金属催化剂已被探索应用于各种类芬顿反应中,如铈、铬、钴、铜、锰、钌、多金属氧盐酸和金纳米材料等。这些金属物种可以将H2O2分解为·OH,我们将讨论它们降解有机物时的氧化还原特征。虽然其活化机制被催化剂的性质所影响,但可以通过其金属络合的配体得到有效的调控。 在所有稀土元素中,铈是唯一可通过类芬顿机制活化H2O2的金属,由于其4f26S2的构造,铈有Ce3+和Ce4+两种价态。Ce3+是强还原剂,在水溶液中易在电极上氧化为Ce4+,而Ce4+在酸性条件下是强氧化剂和强络合剂,因此Ce3+/Ce4+随溶液中酸度和酸根性质变化而变化。在一定条件下,Ce3+和Ce4+两价态之间可自由循环,可能会出现氧空位的现象[21],而氧空位的发生是CeO2催化氧化有机物的前提,氧空位的方程式如式(1)。 (1) 它不仅具有极强的氧化-还原性能,而且还具有优异的存储氧-释放氧性能。而杂质的引入会造成晶格氧缺陷,使其催化活性得到显著提高。因此,CeO2具有较高的氧流动性及优异的催化性能,是所有铈化合物中最受欢迎的选择。 Ce3+/Ce4+的氧化还原反应经常使用CeO2作为非均相类芬顿催化剂氧化有机污染物。在H2O2存在的情况下,Heckert等[22]证实了HO·的产生,类似于Fe2+/H2O2芬顿体系,如式(2)和(3)。但和Fe2+/H2O2体系不同的是,CeO2可在中性条件下降解有机物,Xing等[23]在pH=6~9下利用CeO2光催化降解环丙沙星,降解效果良好。 (2) (3) 裸露的氧化铈不能进行类芬顿反应生成HO·,且Ce3+/Ce4+媒质的氧化能力及再生效率与酸介质密切要关,所以常用高氯酸、硝酸、硫酸或甲磺酸预处理CeO2[24]。而使用最多的是用硫酸预处理CeO2,Yongchuan Wang等[25]认为由于H2O2与表面Ce3+位点的过度络合,所生成的过量活性表面过氧化物(即≡Ce(Ⅲ)—O2H-)可在酸性条件下分解为HO·。除了使用酸处理,候杰等[26]对合成的氧化铈进行氨基修饰,介孔氧化铈经氨基功能化后对酸性红14、偶氮染料AO7和RhB有显著的光催化活性,降解效果良好。 但是稀土元素铈有着极强的细胞毒性,对环境和人类都有危害,所以在实际应用时,要确保氧化铈的催化稳定性和后处理安全性。 Cr(Ⅵ)还原生成Cr(Ⅴ)或Cr(Ⅳ)反应中间体,并与H2O2发生一系列反应生成HO·,如式(4)~式(6)。 (4) (5) (6) 与Cr(Ⅵ)相比,Cr(Ⅲ)与H2O2有着强烈的pH依赖性。在pH≤3时,Cr(Ⅲ)以[Cr(H2O)6]3+的形式与H2O2完全不反应,但在中性pH时,其反应活性和HO·产率最高[29]。类似于Cr(Ⅵ)/H2O2体系,HO·的生成也是个连续的芬顿反应,但Cr(Ⅵ)作为产生的最终价态,如式(7)。 (7) 尽管Cr(Ⅲ)和Cr(Ⅵ)都会与H2O2发生芬顿反应,但可通过pH值严格控制氧化还原途径。通过控制溶液pH值,Cr(Ⅲ)会氧化成Cr(Ⅵ)再通过Cr(Ⅵ)还原再生,而且每次氧化还原转化都伴随着HO·的生成。所以,所有含铬废水都可通过控制H2O2投加量和简单的pH调节,得到有效处理。 (8) (9) 以Co2+为活性金属的有机络合物具有较独特及优异的反应性能,但其氧化还原电位(φCo2+/Co3+=1.82 eV)过高,在正常情况下,不允许下式(10)的发生。 Co3+Lm+OH-+·OH(Lm为配体) (10) Co2+作为非均相芬顿催化剂可以在较宽的pH范围内进行。但是它还存在许多弊端:(1)与铁基芬顿体系相比,H2O2的化学计量需求较高;(2)固定后的络合物浸出过量(>25%);(3)钴的环境毒性大,如果含钴废水直接排放到环境中,将会对人产生巨大危害,所以需确保后处理的安全性。 相较于Fe2+/Fe3+,Cu+和Cu2+表现出更强的氧化还原能力,都可与H2O2发生反应,如式(11)和(12)。 (11) (12) 更重要的是,Cu2+/H2O2类芬顿体系比Fe3+/H2O2表现出更宽的酸碱度工作范围。在近中性或中性的水溶液中,基于Cu2+的芬顿催化剂都能有效地生成HO·,用于各种有机污染物的氧化。与铁基催化剂不同的是,Cu2+络合物不会阻止芬顿反应,实现有机污染物的完全矿化。Cu2+/H2O2除了产生经典还原过程的HO·外,还可直接攻击络合于催化剂表面的有机自由基加合物进而生成一个羟基化产物和另一个·OH,避免了H2O2的无效分解,提高其利用率[34]。 Fe2+与H2O2的反应在酸性溶液中几乎不受氧浓度的影响,但是Cu+与H2O2的反应会被分子氧限制,如式(13)。因为Cu+在酸性和近中性条件下被氧化为Cu2+,降低了Cu+与H2O2反应的有效性。使用Cu2+催化剂的芬顿体系需要更多的H2O2,以补偿HO·的清除作用。但大量的H2O2不仅增加了处理成本,还降低了处理效率。 (13) 为了提高H2O2的利用率,Lai Lyu等[35]用Cu和g-C3N4表面羟基络合,并研究其H2O2的电子转移路径,发现阳离子-π所诱发的电子极化使H2O2的利用率高达90%以上。徐苏倩等[36]制备的Cu-Al2O3-g-C3N4类芬顿催化剂使得 H2O2以其亲电端吸附在富电子Cu中心上,促进了H2O2被还原而不是被氧化,使得H2O2最大限度的被利用,利用率高达72.3%。 锰(Mn)的氧化态范围是0~+7价,虽然Mn6+和Mn7+的价态比较稳定,但只有Mn2+、Mn3+和Mn4+这三种氧化态具有催化意义。锰在水生环境中仅以Mn2+或Mn4+的形式存在,Mn4+可轻易氧化还原为Mn2+,所以MnO2是强大的氧化剂,如式(14),其中S为有机/无机底物。 (14) 为了提高其催化性能,可把MnO2负载在载体上,Li Lu等[37]将MnO2负载在石墨烯纳米片上,将此应用于甲醛的催化。与单纯的MnO2相比,其石墨烯-MnO2杂化纳米片的构造可以使MnO2暴露更多的活性位点,加速其电荷传速,显著提高了降解效率。 锰氧化物作为非均相芬顿催化剂提供了许多优势,例如(a)可在pH近为中性条件下达到降解最佳性能;(b)可根据不同情况而选择性形成活性氧物种;(c)构造不同的锰天然化合物轻松易得,因此在实际应用中很有前景。 在铂族金属中,钌(Ru)是唯一可在H2O2存在下表现出类芬顿的活性元素。钌的氧化态范围是0~+8价,但是目前只发现了二价(Ru2+),三价(Ru3+)和四价(Ru4+)的氧化态。 Abdoulaye Thiam等[40]利用RuO2阳极对含呋喃进行芬顿反应,具体反应见式(15)~式(18)。 (15) (16) (17) (18) Rokhina等[41]使用多孔RuI3粉末作为H2O2的类芬顿催化剂来氧化苯酚。类似于Fe3+/ H2O2芬顿体系,Ru3+还原原位生成Ru2+可引发H2O2分解成·OH,如式(19)。 Ru2++·OH+H+ (19) 金属钌配合物具有热力学稳定性好、激发态反应活性高和使用寿命长等优点,所以在催化降解有机物方面上,其有着极大的应用价值。顾芷萌等[42]用三嗪环类衍生物为配体,以RuCl3为金属源,合成了Ru(L1)Cl3、[Ru(L2)2]·Cl3和[Ru(L2)2]·(H2BTC)·(HBTC)·H2O三种配合物,配合物导致电子云密度发生变化使得电子-空穴对分离,并在H2O2的作用下生成H+和·O2-自由基,对有机染料降解效果显著提高。Debabrata Chatterjee利用RuⅢ(EDTA)/H2O2体系分别对含硫化合物(硫脲和半胱氨酸)[43]、偶氮染料酸性橙Ⅱ[44]进行了降解,在H2O2的存在下,RuⅢ(EDTA)先反应生成[Ru3+(EDTA)(OOH)]2-,其再继续生成HO·降解有机物,如式(20)。 (20) [Ru4+(EDTA)(OH)]-能在类芬顿反应中继续分解H2O2,生成Ru5+氧化态和HO·。 钌催化剂不仅有着高稳定性,还可在中性或偏碱性pH下达到最佳的降解效果,这是最具有优势的。但是钌元素稀有且昂贵,所以为充分利用好钌元素,还得发掘其他改进方法,以达到利用更少钌元素获得更高催化性能的目的。 多金属氧酸盐(Polyoxometalates,简称POMs),是由最高氧化态的过渡金属和氧原子共价形成的无机纳米簇。金属原子通常是第五副族或第六副族的过渡元素,如钼、钒、钽、铌、钨元素。由于其化学结构和元素组成的广泛性和多样性,特制的POMs催化剂广泛应用于催化材料、磁性材料、光电材料、药物等领域。研究者将POMs的氧化能力与H2O2的催化分解相结合,已被广泛用于烯烃的环氧化、醇和苯酚的氧化和乙二醇的转化。 在可见光照射下,通过两种不同的方法实现了有机染料的氧化:(1)POM的光激发,以及从激发态的POM电子转移到H2O2,如式(21)~(23)。(2)染料敏化导致POM单电子还原,随后与H2O2反应形成过氧络合物[45],如式(24)~(26)。其染料氧化直接通过球内氧的转移进行,而不是生成游离HO·降解有机染料。 (21) (22) (23) Dye·+—POM- (24) Dye·+—POM—OOH (25) (26) 有着强水溶性的POMs适合作为均相催化剂,但高溶解性导致其难回收,不适合应用于工程中。研究者们试图对均相催化剂进行改性修饰变成多相催化剂,最常用的是负载型催化剂。Simon Lukato等[47]在多金属氧酸盐上负载Ag-Cu金属制备成Ag-Cu/POM催化剂,用来催化降解苯甲醇,Ag-Cu/POM催化剂稳定性高,没有金属浸出和失活的现象,其多相性质便于回收,重复利用率高。 POMs作为芬顿催化剂的主要优势是具有结构可调控的催化/光化学反应活性,以及较高的化学稳定性和可重复利用率。 近年来,纳米金(AuNPs)由于具有大比表面积,良好的光学特性、尺寸效应和量子效应等特征,在传感、催化、合成、医药等领域受到了广泛关注。 (27) (28) 金基催化剂有着很高的催化活性,H2O2中大约有79%可以转化成HO·,Sergio Navalon等[49]研究出只需用0.5 mg/L的金可降解75 g/L的苯酚,其高催化活性可能是因为Au是惰性材料,在各种反应条件下不太可能浸出,但也有可能是因为Au对H2O2分解的灵活选择性。Sergio Navalon等[50]在金刚石上负载Au形成Au/OH-npD催化剂,在黑暗条件下Au/OH-npD催化剂不发生催化反应,但在单色光或太阳光下,都能促进此催化剂的催化活性,可归因于光源促进了电子从金转移到H2O2。但金含量和粒径都会对光化学过程有显著的影响,为了防止热弛豫引起的粒子团聚和生长的现象,Au的负载量要低于1%,且粒径尽可能的小,至少低于10 nm。 虽然金基催化剂有着很高的催化活性,但它的成本远比其他金属要贵,所以在实际应用中的机会不大,今后的研究方向应注重于提高其重复利用率上。 基于芬顿的高级氧化技术主要是用铁基催化剂,但是铁基催化芬顿反应需要严格的酸性环境,酸化反应后需要中和处理。铁基催化反应还容易形成铁污泥,造成二次污染,因而对反应设备要求高,限制了其在实际工程中的应用。 在均相和多相体系下,非铁催化剂具有多种氧化状态和高稳定性的优势,即使在中性或偏中性条件下也能分解H2O2生成HO·,因而受到人们的青睐。但是每种有色金属催化剂也存在缺陷。铈、铬和钴等可溶物质具有细胞毒性,只能在可控条件下应用;多相锰氧化物具有环境友好、天然丰度高等优点,但它存在物化性质和反应机理较复杂的缺点;钌络合物、多金属氧盐酸和金纳米材料是非常稳定的催化剂,但应用成本较高;铜基催化剂需要过量的H2O2,并且需严格的厌氧条件。 未来为了建立实际可行、环境可持续性的非铁芬顿体系有待于在以下方面进行展开: (1)继续寻求高稳定性、零金属浸出、无二次污染的催化剂。可以直接在催化剂上做出改进,比如向体系中加入氧化还原介质、络合剂等化学试剂,大多络合后的催化剂可以达到低金属浸出率和高稳定性的目的。但需进一步探讨更好、更有效的改进方法。 (2)发展非铁基强化类芬顿技术。效仿铁剂催化剂类芬顿技术,向非铁基催化反应体系施加紫外光、电场或超声波等,从而加强对H2O2的活化激发,提高反应效率。 (3)探讨非铁基芬顿/类芬顿技术和其他工艺的组合/耦合技术,优化处理系统的运行条件,提高处理效果。如芬顿/类芬顿法可以通过激发H2O2产生HO·实现对难降解有机物分解,提高污废水的可生化性,因此,可以考虑与生物处理单元结合等。 相信通过研究者们对类芬顿技术的不断探索和优化,将达到高效率、低成本和高环境相容性的目的,进而投入使用到大规模的实际工程中。
1.3 铁基催化剂的利弊
2 非铁催化剂的发展
2.1 铈

2.2 铬
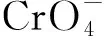
2.3 钴
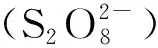
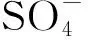
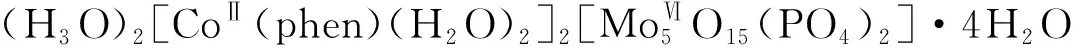
2.4 铜
2.5 锰

2.6 钌
2.7 多金属氧酸盐
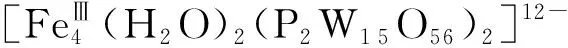
2.8 金纳米材料

3 结论和展望
