Self-assemblies of TTF derivatives with fluorinated phenyls and pyridine group
2022-06-18JinghuFngXioyngZhuWendiLuoJinxunShiLejiWngBinTuQingdoZengXunwenXio
Jinghu Fng, Xioyng Zhu, Wendi Luo, Jinxun Shi, Leji Wng, Bin Tu,Qingdo Zeng,*, Xunwen Xio
a College of Material Science and Chemical Engineering, Ningbo University of Technology, Ningbo 315211, China
b CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology (NCNST), Beijing 100190, China
c Zhejiang Pharmaceutical College, Ningbo 315100, China
ABSTRACT The self-assembly characteristics of tetrathiafulvalene (TTF) derivatives molecules 1–3 at the 1-phenyloctane/HOPG (HOPG = highly oriented pyrolytic graphite) interface had been carefully studied by scanning tunneling microscopy (STM) method.The number of F atoms on the phenyl group had significantly affected the self-assembly structures.High-resolution STM images make clear the different assembly structures between the molecules 1–3, which attribute to the different F atom numbers and pyridine group in the molecule.Density functional theory (DFT) calculations have been performed to reveal the formation mechanism.
Keywords:TTF Self-assembly STM DFT calculation Hydrogen bonds
In view of the excellent electron-donating ability and reversible redox features, tetrathiafulvalene derivatives (TTFs) were extensively utilized as donor units in the molecular material, especially in the organic conductor/superconductor materials [1,2].The oxidation potential of TTFs could be modified by peripheral substitution on the TTF core.Previous research results made clear that the stacking pattern of TTFs could also be influenced by the substituent groups.Tetrathiafulvalene derivatives (TTFs) which were modified by peripheral arylation showed many advantages to get organic optoelectronic materials [3,4].Moreover, phenyl and pyridine units are the most common peripheral aryls unit.The different peripheral aryls units in the TTF core play a key role in constructing different arrangements in the condensed matter based on such donor molecule [5–7].
We previously reported the self-assemblies of TTF-pyridyl derivatives at the liquid/solid interface with the scanning tunneling microscope (STM) method.With one pyridine group connected with the TTF core, 4-pyridyl-(ethylenedithio)TTF (EDTTF)could form a brand network with the help of 1,3,5-tris(10-carboxydecyloxy)-benzene at room temperature, which finally transforms into a more stable line structure [8].When two pyridine groups were directly lined with TTF core, the para-pyridine-TTF and meta-pyridine-TTF molecules could both assemble into a regular linear structure at the heptanoic acid (HA)/HOPG interface.And also, by co-assembling with the TCDB (TCDB = 1,3,5-tris(10-carboxydecyloxy)-benzene) molecule, these two TTF derivatives could disturb the H-bonding network of TCDB, and form new co-assembled structures [9].More, the directionality of hydrogen bonds tends to facilitate the arrangement of neighboring molecules and surface structures controlled by hydrogen bonds are reported recently [10–13].These results indicate that the assembled mode of the TTF derivatives could be affected by changing the substituent on the TTF molecule.Therefore the self-assemblies of the TTFs are still worth further exploring.
In this paper, we modify the TTF molecule with fluorine (Fluorine element has the highest electronegativity) which could offer two obvious advantages: (1) fluorination could reduce both the HOMO and the LUMO energy levels of these organic optoelectronic molecules, so that the p-type organic semiconductor may be converted into the n-type one; (2) fluorine-involved intermolecular interactions play an important role in the crystal engineering of such kind molecule, leading to the assembly structure more orderly, consequently enhancing the charge carrier mobility of TTF derivatives [14–16].Herein we design molecules 1–3, which fluorinated phenyls and/or pyridyl group linked with the TTF core, as shown in Scheme 1.The difference between molecules 1–3 is the F atom number on the aromatic group of terminal groups.The selfassembled structures of 1–3 were studied by the STM method for having absolute predominance in revealing the assembled structure at the atomic level [17].Moreover, density functional theory(DFT) calculations have been executed to resolve the possible formation mechanism of their self-assembly structures.

Fig.1.STM images of molecule 1 assembly structure at the HOPG/1-phenyloctane interface: (a) Large scale; (b) high resolution.Tunneling conditions: Iset = 198.4 pA,Vbias = 499.9 mV.
Molecules 1–3 were separately dissolved in 1-phenyloctane,and each concentration is less than 10-4mol/L.Then the selfassembly structures were prepared by depositing a droplet of the corresponding solution (0.1 μL) onto the freshly cleaved highly oriented pyrolytic graphite (HOPG) surface, respectively.Afterwards, the self-assembly structures were observed at the HOPG/1-phenyloctane interface by using the STM method under ambient conditions.More, the detailed synthesis of compounds 1–3 is shown in Fig.S1 (Supporting information) and the detail of STM investigation and theoretical calculations are shown in Fig.S2(Supporting information).
First, the assembly structure of molecule 1 at the 1-phenyloctane/HOPG interface was characterized by using STM.The large-scale nanostructure of molecule 1 was shown in Fig.1a, and the high-resolution image of the self-assembled structure was revealed in Fig.1b.Fig 2 shows the proposed corresponding molecular model based on the DFT calculation.From Fig.1b, three different assembly structures (A–C) of compound 1 were formed in the same area at the same time.More detailed structure was seen in high resolution STM image, as shown in Fig.1b.Type A is dimer; type B contained six molecules; and type C contained eight molecules.The fluorinated phenyls and pyridyl groups in the molecule are in thecisconfigurations.As a result, molecule 1 adopts a U-shaped structure [18].Type A is a dimer which was formed by two molecules with a tail to tail mode.Type B was formed by six molecules with head to head and tail to tail mode.And C was formed by eight molecules with head to head, tail to tail, and head to tail mode as shown in Fig.2.The width (W) of the bright spots is 0.8 ± 0.1 nm which is in accordance with the size of TTF.Therefore we could deduce that each bright spot represents one molecule 1.Each bright rectangle spot corresponds to the conjugated backbone of the TTF molecules due to the high electron cloud density.In type A, the two molecules in the dimer can interact with each other through C–H···F and C–H···N hydrogen bond between fluorophenyl and pyridine groups in 1 (marked by the red dashed circle for C–H···F and black dashed circle for C–H···N in Fig.2a).And it was indicated by a rectangular box marked A in Fig.1b.And the measured unit cell parameters for type A area= 1.3 nm ± 0.1 nm,b= 2.0 nm ± 0.1 nm,α= 90°± 2°.In type B, these are C–H···N and C–H···F hydrogen bonds from two pyridine group with a tail to tail mode, which marked by the red dashed circle for C–H···F and black dashed circle for C–H···N in Fig.2b and F···F halogen-bond interaction (marked by the violetdashed circle for F···F in Fig.2b) from two 4-fluorophenyls with head to head mode.The measured unit cell parameters for type B area= 4.5 nm ± 0.1 nm,b= 3.5 nm ± 0.1 nm,α= 130°±2°.For type C, there are C–H···N, C–H···F and C-F···F interactions in this ring of eight molecules (marked by the red dashed circle for C–H···F, black dashed circle for C–H···N and the violet dashed circle for F···F in Fig.2c).The measured unit cell parameters for type C area= 6.1 nm ± 0.1 nm,b= 3.0 nm ± 0.1 nm,α= 120°± 2°,respectively.
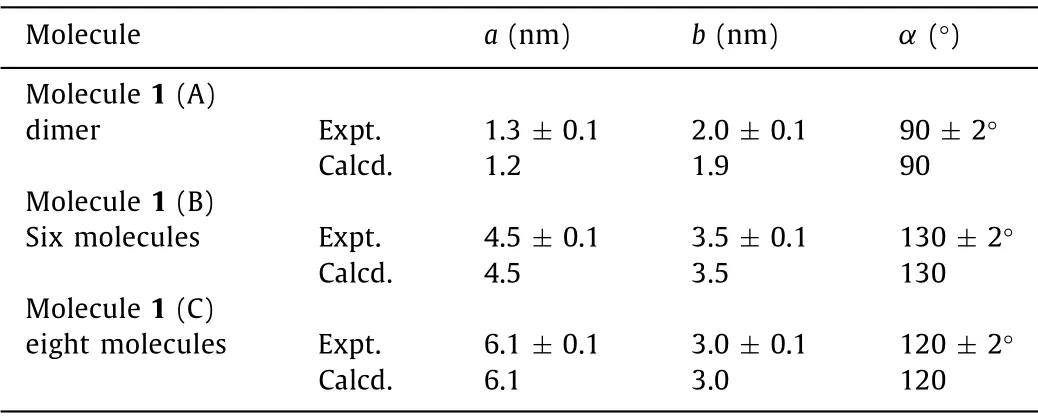
Table 1 Experimental (Expt.) and calculated (Calcd..) unit cell parameters of the selfassembly of molecule 1 on the HOPG surface.
Therefore, we could speculate that the intermolecular halogen bond, hydrogen bond interaction in TTF core play an important role in the self-assembly process.There is little difference between these intermolecular interactions.On the basis of STM observations, molecule 1 could form three types of self-assembly structures.Fig.2 showed the corresponding molecular model proposed by the DFT calculations.The calculated lattice parameters for the self-assembly structures of molecule 1 are summarized in Tables 1 and 2.Combined with the DFT result, the total energy per unit area of 1 of A–C was equal to -0.261, -0.175 and -0.149 kcal mol-1-2, respectively.There is little difference of the lowest energy of these three type modes.As a result, three self-assembly structures of molecule 1 were formed.It is notable that the total energy per unit area of type A is the lowest, indicating that the type A is the most energetically favourable pattern of these three type structures.
Then we investigated the effect of different F atoms of TTF derivatives on their assembly behavior.In molecule 2, two H atoms of the benzene ring were replaced by two F atoms.Compared with molecule 1, only one more H atom of benzene ring was replaced by an F atom in molecule 2.However, the assembly of 2 at 1-phenyloctane/HOPG interface showed much difference with molecule 1.As seen in Fig.3, molecule 2 formed only one arrangement in a large-scale order.The width (W) of the bright stick is 0.8 ± 0.1 nm, which is in accordance with the width of molecule 2.Therefore, the bright structure could be assigned to only one molecule 2.The measured unit cell parameters area= 3.9 nm ± 0.1 nm,b= 1.1 nm ± 0.1 nm,α= 80° ± 2°, and this was indicated by a white rectangular box in Fig.3b.On the basis of STM observations, a suggested molecular model is proposed, as shown in Fig.3c.Molecule 2 also adopts thecisconfigurations.The up and down configurations of 2 are column arrangements.For the assembled structure of 2, two adjacent 2 molecules adopt the head to head and tail to tail mode.Molecule 2 contains two F atoms;two pairs of C–H···F hydrogen bonds could be formed (marked by the red dashed circle for C–H···F in Fig.3c).Moreover, C–H···N hydrogen bonds could also be formed with the help of pyridine group(marked by the black dashed circle for C–H···N in Fig.3c).From the STM images, we can estimate that fluorine atoms and pyridine group participate in assembly behavior and the intermolecular interactions are mainly C–H···F and C–H···N hydrogen bonds.Therefore, the assembly structure of 2 showed single regular structures.Moreover, almost no defects were found in large areas.On the basis of STM observations, Fig.3c shows the corresponding molecular model proposed by the DFT calculations.The calculated lattice parameters for the self-assembly structures of molecule 2 are summarized in Table 3.The intermolecular C–H···F and C–H···N hydrogen bonds are relatively strong.As a result, molecule 2 interacted with each other by the head to head mode through the hydrogen bonds of the terminal fluorobenzene and pyridine group.
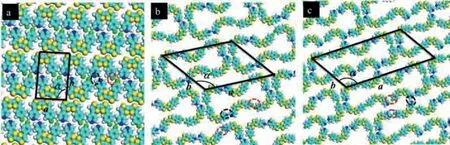
Fig.2.Three simulated molecular packing structures of 1, (a) for type A; (b) for type B, (c) for type C.

Table 2 Total energies and energies per unit area of self-assembly of molecule 1 on the HOPG surface.a

Fig.3.STM images of molecule 2 assembly structure at the HOPG/1-phenyloctane interface: (a) Large scale; (b) high resolution.Tunneling conditions: Iset = 212.4 pA,Vbias = 489.8 mV.(c) The simulated molecular packing structure.

Table 3 Experimental (Expt.) and calculated (Calcd.) unit cell parameters of self-assemblies of molecules 2 and 3 on the HOPG surface.
In order to further investigate the effect of the number of F atoms on the self-assembly of TTF core, molecule 3 was designed.In molecule 3, two difluorobenzene groups were directly connected with TTF core and no pyridine group appeared in molecule 3.At the HOPG/1-phenyloctan interface, molecule 3 forms a grid selfassembly structure, which is different from that of molecule 1 or 2.As seen from the Fig.4, the width (W) of the bright spot is 0.7 ± 0.1 nm, which is slightly less than that of molecule 1 or 2.The measured unit cell parameters area= 3.3 nm ± 0.1 nm,b= 1.4 nm ± 0.1 nm,α= 55° ± 2°, and this was indicated by a white rectangular box in Fig.4b.On the basis of STM observations, a suggested molecular model is proposed, as shown in Fig.4c.Molecule 3 also adopts thecisconfigurations.There are two different spots on the STM images, which could be identified as up or down configuration of molecule 3.In these systems, the main molecular interaction is C–H···F hydrogen bond (marked by the red dashed circle for C–H···F in Fig.4c).As the number of fluorine atoms in the phenyl increased, the interaction related with fluorine atoms was also increasing.Four pairs of C–H···F hydrogen bonds could be formed between the adjacent molecules because there are four F atoms in the molecule.And the intermolecular interactions were stronger than those of molecule 2, the density of the whole assembly structure of 3 was bigger than that of 2.
Table 3 lists the calculated unit cell parameters of molecules 2 and 3 assembly systems, which agree well with the corresponding experimental results.The interaction energies of molecules 2 and 3 self-assemblies were presented in Table 4 and the lower energy indicates the stronger interaction herein.It can be observed that the total energy per unit area of molecule 3 (-0.115 kcal mol-1-2) is lower than that of molecule 2 (-0.067 kcal mol-1-2), which is largely due to the extra C–H···F hydrogen bond interactions between difluorobenzene groups of molecule 3.Except the interaction between self-assembled molecules 2 and 3, the interaction between molecules and HOPG also plays an important role in the surface assembly.As shown in the third column in Table 4, the interaction energy between molecule 3 and substrate(-39.79 kcal/mol) is lower than that of molecule 2 and substrate(-26.55 kcal/mol), which caused the different interaction between TTF core and graphite substrate.Compared with the intermolecular interaction energies of molecules 1–3, the interaction energies between molecules and HOPG are much lower.These results may indicate that the absorption between molecules and HOPG substrate is quite strong.

Fig.4.STM images of molecule 3 assembly structure at the HOPG/1-phenyloctane interface: (a) Large scale; (b) high resolution.Tunneling conditions: Iset = 230.0 pA,Vbias = 499.9 mV.(c) The simulated molecular packing structure.
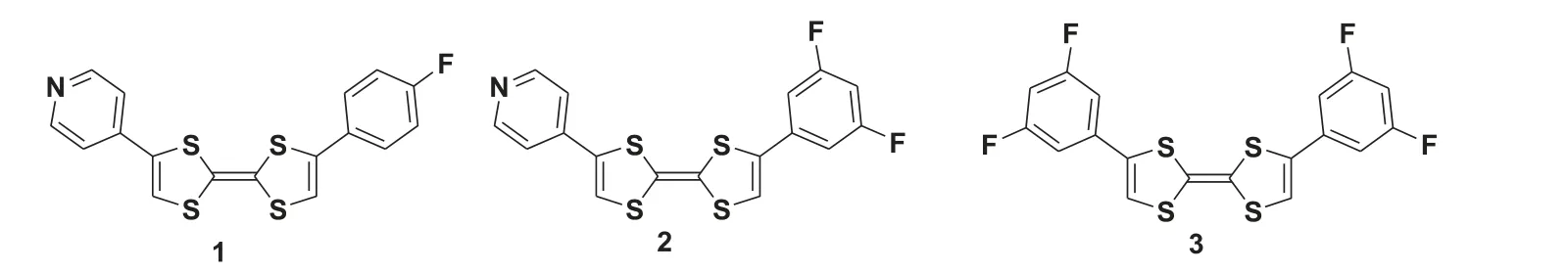
Scheme 1.Chemical structures of TTF-based molecules.
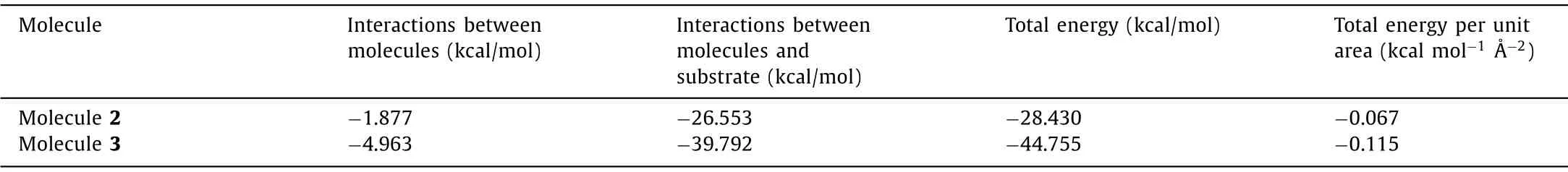
Table 4 Total energies and energies per unit area of self-assemblies of molecules 2 and 3 on the HOPG surface.a
From the experiment results, we could conclude that the three molecules 1–3 assemble into the linear patterns at the HOPG/1-phenyloctane interface.However, the different numbers of F atoms on the molecule cause the different assembly structures.As for the self-assembly of molecule 1, the self-assembly structure is formed by the help of C-F···H, C–N···H hydrogen bond and F···F halogen bond.As for the self-assembly of molecule 2, the linear pattern is formed by the C-F···H and C–N···H intermolecular hydrogenbond interaction.As for 3, the assembly pattern is formed with the help of the C-F···H intermolecular hydrogen-bond interaction.Due to no alkyl chains in molecules 1–3, the interactions between molecules and substrate may come from theπ-πstacking interaction between TTF core and graphite substrate.Therefore the peripheral substitution or chemical modification of the TTF core is important for the self-assembly on the graphite substrate.Combined with the DFT result, the total energy of one molecule 3(-44.75 kcal/mol) is much lower than that of molecule 2 (-28.43 kcal/mol) and molecule 1 (type A: -59.559/2 = -29.78; type B:-211.731/6 = -35.28; type C: -236.486/8 = -29.56 kcal/mol).As a result, a closest molecular arrangement and the best thermodynamic stability of molecule 3 are formed among molecules 1–3.
Taking advantage of DFT calculation, the precise self-assembled structures of molecules 1–3 based on the observed STM image were understood.The calculated parameters are in good agreement with the experimental data.The molecules 1–3 make use of the F···F, C–N···H and C–F···H interaction to form self-assembly structure.Due to the different number of F atoms on the fluorinated phenyls and pyridine group which directly connected to the TTF core, molecules 1–3 showed different self-assembly characteristics and formed different 2D morphologies of nanostructures.As a result, the F···F, C–N···H and C–F···H interactions could play a vital role in stabilizing such molecules on the surface, which helps us to predict and construct new structures of functional molecules.
In summary, the self-organizing behavior of fluorobenzene-TTF derivative molecules 1–3 without long alkyl chains had been studied by STM method for the first time.At the HOPG/1-phenyloctan interface, molecule 1 containing fluorobenzene and pyridine group could form three kinds of assembly structure.With one difluorobenzene group and one pyridine group, molecule 2 shows single regular structures with few defects in the self-assembly area.Moreover, with two difluorobenzene groups connected to the TTF core, molecule 3 showed a more compact arrangement compared with molecule 2.These results showed that the packing of TTF cores could be effectively controlled by the different number of F atoms on the substitution on the TTF molecule.This new phenomenon may make us understand the self-assembly of TTF at interfaces more clearly.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported NSF of Zhejiang Province of China(Nos.Y20B020032, LY18B020016), the National Natural Science Foundation of China (Nos.21773041, 21805144 and 21972031) and the Strategic Priority Research Program of Chinese Academy of Sciences (No.XDB36000000).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.08.030.
杂志排行

Chinese Chemical Letters的其它文章
- Comment on “Acid-induced tunable white light emission based on triphenylamine derivatives”
- Strategies for efficient photothermal therapy at mild temperatures:Progresses and challenges
- Liposome-based delivery of biological drugs
- Macrophage-targeted nanomedicine for chronic diseases immunotherapy
- Advances, opportunities, and challenge for full-color emissive carbon dots
- Fluorine-containing agrochemicals in the last decade and approaches for fluorine incorporation