簸箕柳R2R3-MYB转录因子家族全基因组分析
2022-06-17王岚春沈方圆欧阳丹
王岚春, 沈方圆, 欧阳丹, 李 校
(四川大学生命科学学院 生物资源与生态环境教育部重点实验室, 成都 610064)
1 引 言
生物体会通过基因的表达和沉默来调控自身的生长发育,其调控过程是相互独立而又相互依存的.这种调控可能发生在基因表达过程中的任意一个阶段,如 DNA 的转录过程,mRNA的加工过程,以及 mRNA的翻译过程等,且调控的发生需要各种酶和调节蛋白的相互配合[1].其中转录因子参与DNA转录起始过程来影响基因的表达.转录因子通常被定义为具有序列特异性的DNA结合并能够激活或抑制转录的蛋白质[2].基因表达在转录水平上的调控影响或控制着细胞或有机体中的许多生物学过程,如细胞周期的进展、代谢和生理平衡以及对环境的反应.
漫长的自然选择与进化过程当中,植物形成了其特有的基因表达和调控机制,以适应不断改变的生存环境.转录因子在这一过程中起到了很大的作用.MYB转录因子广泛分布于所有真核生物中,是植物界最大的转录因子家族之一[1].大多数MYB蛋白起着转录因子的作用,具有不同数量的MYB结构域重复序列,赋予它们结合DNA的能力[1].MYB蛋白是控制发育、代谢和对生物和非生物胁迫反应的调控网络中的关键因子[3].
MYB转录因子(TFs)是一组由1到4个MYB重复序列定义的全真核转录因子.50个氨基酸组成三个α螺旋,每个重复序列的第二个和第三个螺旋形成一个螺旋-旋转-螺旋(HTH)结构,其中有三个间距规则的色氨酸(或疏水)残基,形成疏水核心.每个重复序列的第三个螺旋是与DNA直接接触的DNA识别螺旋[4]. HTH与启动子中的调节元件相互作用,而C-末端区域负责与真核转录机制的其他成分建立蛋白质-蛋白质相互作用[5].在DNA接触过程中,两个MYB重复序列紧密地堆积在主沟中,使两个识别螺旋协同结合到特定的DNA识别序列基序上.MYB-DNA结合域的长度为100~160个残基,这取决于N-末端区域不完全重复(称为R)的数量.根据MYB重复数和MYB重复序列的特性,MYB蛋白质一般分为MYB-related、R2R3-MYB、R1R2R3-MYB和4R-MYB蛋白质[1].在这四种类型中,R2R3MYB是高等植物特有的,在大多数植物中数量占优势,其特点是存在保守的MYB结构域和高度可变的C-末端区域[6, 7].MYB TFs是植物中最大的TF类之一,该类TF的规模主要归因于R2R3-MYB TF家族的迅速扩张[8].先前的研究表明, R2R3-MYB TFs的数量随着绿色植物进化过程而增加[9].
越来越多的证据表明,R2R3MYB转录因子参与植物许多生理生化过程,如叶毛状体分化[10]、次生壁形成[11]、花药和花粉发育[12, 13]、腋生分生组织形成[14],二次代谢的调节包括类黄酮[15]、花青素[16]和木质素[17].除此之外,R2R3MYB家族成员还参与植物对各种非生物和生物胁迫的防御和响应[18-20],并在调节植物对包括吲哚乙酸[21]、脱落酸[21, 22]、赤霉素[23, 24]、乙烯、水杨酸和茉莉酸[24]等植物激素线索的反应中发挥了作用,以及一些环境信号响应,如水可用性[25],光[26]和营养元素[27].
随着多种植物的全基因组测序完成,实验技术及手段的进步,使得科研人员可以对 R2R3-MYB 基因家族在内的基因资源进行更为广泛和深入的研究.对于R2R3-MYB转录因子的生物功能研究也有了很大的进展,越来越多的R2R3-MYB转录因子的功能被揭示.虽然在一些物种中已经对该家族进行了全基因组分析,但对簸箕柳(Salixsuchowensis)的R2R3-MYB基因知之甚少.簸箕柳属于杨柳科中的柳属,筐柳组,因其枝条强韧,经常被用来编制筐篮等农具,是一种有发展前途的经济植物.除此之外,簸箕柳也可用作固砂树种、河堤边的防浪林树种,具有一定的生态价值.对簸箕柳进行R2R3-MYB基因的全基因组鉴定,且对其进行该家族的多样性、进化和其功能研究,有助于了解该蛋白的扩展过程和机制以及随后该蛋白的亚功能化和新功能化机制.进一步探索R2R3-MYB参与簸箕柳生命生长过程的途径、方式、调控机制还可以为改良作物遗传及抗逆提供理论依据及支持,也可以直接作为基因资源加以利用.更为详细的关于系统发育分析、辅助基序和DNA结合特异性的发现也会为深入了解植物R2R3-MYB转录因子的进化史提供线索.除此之外,因簸箕柳枝条强劲、十分耐涝,是河堤边防浪林树种的良好选择,其具有耐性和韧性的枝条在水波中起到了很好的缓冲作用,但洪水或者大风天过后往往造成树木的倒伏,研究簸箕柳的抗重力刺激响应为后期提升簸箕柳的抗倒伏性能提供了一定的理论基础.
2 材料与方法
2.1 材 料
从PopGenIE[28]数据库(http://popgenie.org/)下载簸箕柳的基因组、蛋白质序列和注释文件;从转录因子数据库PlantTFDB[29]v5.0(http://planttfdb.gao-lab.org/)下载拟南芥MYB转录因子蛋白序列.
2.2 方 法
2.2.1 簸箕柳R2R3-MYB转录因子家族的鉴定及其保守结构域分析 根据Pfam数据库[30](http://pfam.xfam.org/)中MYB转录因子家族保守结构域的HMM文件(PF00249),用HMMER 3.0[31]软件对簸箕柳蛋白质序列进行本地搜索.将拟南芥R2R3-MYB转录因子蛋白序列作为查询序列,对簸箕柳蛋白质数据库进行BLASTP搜索.将簸箕柳蛋白质序列传入iTAK数据库[32](http://itak.feilab.net/cgi-bin/itak/index.cgi)进行线上转录因子家族鉴定.将上述方法得到的蛋白序列整合、去除冗余后,批量搜索Pfam数据库[30]和NCBI-CDD数据库[33](https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi)去除结构域不完整的蛋白序列.将鉴定出的蛋白进行重新命名:SsMYB001-SsMYB158.
将上一步鉴定出的簸箕柳R2R3-MYB转录因子家族的蛋白序列导入ClustalW[34]进行多序列比对,将得到的结果进行统计.利用网络服务器MEME[35](http://meme-suite.org/tools/meme)绘制保守结构域sequence logo图形.
2.2.2 系统发育、基序组成、基因结构分析 将上一步鉴定出来的簸箕柳R2R3-MYB转录因子家族成员的序列导入MAGA-X[36]软件,采用MAGA-X自带的ClustalW进行多序列比对,邻接法(NJ),检验参数(Bootstrap)设置为1000,进行系统发育分析,将得到的结果进行亚组分类.使用网络服务器MEME[35]寻找簸箕柳R2R3-MYB转录因子家族成员的保守基序,预期搜索数量设置为20,允许最小宽度为6,允许最大宽度为50.
使用在线软件iTOL[37](https://itol.embl.de/)进行系统进化分析和基序组成分析可视化.根据簸箕柳注释文件,使用TBtools[38]进行基因结构分析可视化.
2.2.3 亚细胞定位及理化性质分析 使用在线软件CELLO[39](http://cello.life.nctu.edu.tw/)对簸箕柳R2R3-MYB转录因子家族成员进行亚细胞定位预测.采用本地Perl脚本对簸箕柳R2R3-MYB转录因子家族成员的基本理化性质进行分析,如理论等电点(pI)、分子量等.
2.2.4 染色体定位和基因复制分析 采用多重共线性工具包MCScanX[40]对簸箕柳R2R3-MYB转录因子家族成员进行基因复制事件分析,利用TBtools[38]和Circos[41]工具绘制共线性图形.采用kaks_Calculator2.0[42]软件计算基因对的ka/ks值.使用RepeatMasker[43]对簸箕柳基因组进行重复序列鉴定,并统计重复序列和基因密度,基于相对于染色体其他区域的高重复序列丰度和低基因含量,预测了每条染色体的着丝粒位置.
2.2.5 GO注释与基因表达分析 将SsMYBs与NCBI的Nr数据库进行blast比对,结果导入Blast2GO[44],最后使用R语言的GGplot2将注释结果可视化.
从NCBI_SRA数据库(https://www.ncbi.nlm.nih.gov/sra/)下载簸箕柳重力刺激相关RNA-Seq数据(SRA号:SRR9849616、SRR9849619-SRR9849623).运用HISAT2+Stringtie[45, 46]流程计算各基因表达量,再使用DEseq2[47]对两组数据进行差异表达分析,选择logFC绝对值大于1且FDR小于0.01的基因为差异表达基因.
3 结果分析
3.1 簸箕柳R2R3-MYB转录因子家族鉴定及保守结构域分析
在簸箕柳中,经Pfam数据库和NCBI-CDD数据库验证,去除相关结构域不完整的蛋白序列,一共鉴定出158个簸箕柳R2R3-MYB转录因子家族成员.将鉴定出的蛋白进行重新命名:SsMYB001-SsMYB158.
在植物界,R2R3-MYB是MYB蛋白中最大的一个亚群,它含有由两个相邻MYB重复序列组成的高度保守的DNA结合域[48].为了探索SsMYBs(R2R3-MYB)成员结构域的特征,提取了SsMYBs成员的MYB结构域,并对其氨基酸序列进行了多重序列比对,结果可以用来检测SsMYBs蛋白的R2和R3重复序列中每个残基位置的保守性.通过数据统计和MEME可视化得到结构域的一致性序列(图1)和相关sequence logo(图2).结果显示,SsMYBs结构域平均约含有105个氨基酸残基(图2),两个重复序列中插入或缺失频率很低.
根据先前的报道,R2和R3重复序列具有特征性氨基酸,包括一系列分布均匀且高度保守的Trp(W)色氨酸(或疏水氨基酸)残基[7].在158个

图1 簸箕柳R2R3-MYB转录因子家族的保守结构域横坐标为重复序列中氨基酸出现的相对位点,纵坐标为该位点占比最大氨基酸的百分比Fig.1 The conserve domain of S.suchowensis R2R3-MYB proteins
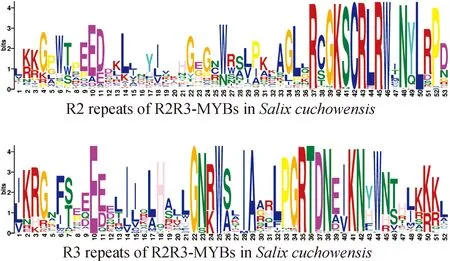
图2 SsMYBs保守结构域sequence logo图Fig.2 The sequence logo of conserve domain of SsMYBs
SsMYBs蛋白中,R2重复序列含有3个色氨酸残基,其分别位于第6、26和46位,这些色氨酸残基形成疏水核心,是植物MYB结构域的典型标志.其中,其中第一个色氨酸残基有部分被组氨酸(H)丝氨酸等残基替代,第二个色氨酸残基有部分被天冬酰胺(N)替代.在R3重复序列中,大多数成员的第一个色氨酸残基(位于6)被苯丙氨酸(F)取代,第二个色氨酸残基(位于25)和第三个色氨酸残基(位于44)在几乎所有SsMYBs中都很好地保存,尤其是存在于所有成员中的第二个色氨酸残基.除了保守的W外,R2重复序列末端还存在十二个高度保守的氨基酸残基:R-37、G-39、K-40、S-41、C-42等,R3重复序列中的E-10、G-22、A-29等也高度保守(图2).如图1所示,SsMYBs结合域中的保守区域主要位于两个R重复序列的第二个和第三个色氨酸残基(每个重复序列中HTH结构域的第三螺旋)之间.SsMYBs结合域中每个R重复序列的第一个和第二个色氨酸残基(第一螺旋)之间的氨基酸序列相对不保守.除此之外,通过sequence logo(图2)可以更直观的看出R3重复片段结构域比R2要更为保守.相比之下,SsMYBs结构域以外的区域在长度和氨基酸组成方面的保守性较差.
3.2 系统发育、基序组成、基因结构分析
通过多重序列比对和Bootstrap分析,研究了SsMYBs转录因子家族成员的系统发育分析(图3).除SsMYB101未能很好地成簇外,其余157个家族成员一共被分为了23个亚组,不同的颜色代表了不同的亚组(图3).基因结构也可以提供进化信息[49].为了深入了解SsMYBs家族的进化,对158个SsMYBs基因的内含子外显子的数目和排列进行了分析.结果表明,同一亚组中的基因通常具有相似的内-外显子结构(图3).SsMYBs成员的基因结构存在显著差异,包括外显子和内含子的数目和相对位置.其中,内含子的数目从0到12不等,大多数基因有两个内含子(65%)或一个内含子(23%).在四个SsMYBs基因中发现了三个内含子:SsMYB069,SsMYB027,SsMYB033,SsMYB053.而在SsMYB059、SsMYB034、SsMYB058和SsMYB046中分别发现了7、10、11和12个内含子.此外,10个SsMYBs基因被发现是无内含子的.同一亚群中同源性最高的成员通常具有相同或平行的外显子/内含子模式,表现出相似的数量、位置和外显子长度.例如,C2中的七个SsMYBs拥有两个外显子和一个内含子、C4中的两个SsMYBs没有内含子、C11中的十个SsMYBs包括三个外显子和两个内含子.但是,在C12和C18中各存在一个例外,SsMYB034、SsMYB059的外显子和内含子的位置以及长度与同组其他成员存在显著差异,成员间的遗传相似性较低.值得注意的是,在每个亚组的末端节点中发现一对或多对高度同源的SsMYBs,这表明这些蛋白质具有相似的功能.
利用MEME程序预测了SsMYBs蛋白的保守基序,共鉴定出20个不同的基序.这些基序被命名为基序1~20,同一亚组的SsMYB成员具有相似的基序结构.值得注意的是,motif14只在C7亚组中存在,motif8、motif10、motif13只在C2亚组中存在且十分保守,motif12只在C6亚组中存在,motif17只在C10亚组中存在,motif17只在C17亚组中存在,motif15只在C18亚组中存在.这些亚组中特有的motif可能有助于其功能分化.在同一亚组的成员,通常具有相似的基序组成和相似的基因结构(图3).
3.3 亚细胞定位及理化性质分析
SsMYBs蛋白的长度在164到1041个氨基酸之间,长度普遍位于200~400 aa,平均长度为332 aa.其中,SsMYB099是最长的蛋白质,含有1041个氨基酸;SsMYB152和SsMYB106是最短的蛋白质,各含有164个氨基酸.SsMYBs的分子量在18.3到117.3 kD之间,平均分子量是37.2 kD.SsMYBs的理论等电点在4.61到10.14之间.158个SsMYBs蛋白呈现不规则的理论等电点特征,75个酸性蛋白(47.5%)和83个碱性蛋白(52.5%).大部分SsMYBs蛋白(87%)的亚细胞定位结果显示其位于细胞核内的.

图 3 簸箕柳R2R3-MYB转录因子家族的保守结构域的系统发育树、motif组成及基因结构
3.4 染色体定位和基因复制分析
片段重复和串联复制是导致植物基因家族扩张的两个主要原因,由同一基因座扩张而来的基因往往具有相似度较高的序列.根据簸箕柳注释信息,对SsMYB基因进行染色体定位分析,有7个基因无法定位到簸箕柳19条染色体上,而其余151个基因则不均匀、无规律地分布于19条染色体上(图4).其中1号染色体上该家族基因数目最多,有15个;11号染色体上的最少,只有一条.SsMYB基因在1、2、3号等染色体有较高的密度.相反,一些大的染色体区域缺乏SsMYB基因,如11号染色体的中央和底部部分、14号染色体的底部.
在SsMYB基因中存在两组串联复制,涉及5个SsMYB基因,分别位于13号染色体和19号染色体.此外,使用MCScanX对SsMYB基因进行片段重复或全基因组重复分析(图5).共鉴定出83个片段重复对,涉及101个SsMYB基因.在这101个SsMYB基因中,58个基因只出现一次记录,43个基因存在于不止一次的片段重复事件中.这些结果表明,约(104)65%的SsMYB基因可能是由重复事件产生的,在簸箕柳MYB基因家族的扩展中起主要作用.
Ka/Ks比值用于估计中性突变、纯化选择和有益突变之间的平衡.计算了串联和片段复制基因对的Ka/Ks比值.结果表明,所有Ka/Ks比值均小于1,表明簸箕柳MYB复制成员在进化过程中可能经历了纯化选择压力.

图 4 簸箕柳R2R3-MYB转录因子家族在染色体上的分布和串联复制事件

图5 簸箕柳R2R3-MYB转录因子家族的片段复制事件
3.5 GO注释
相似的基因在不同物种中,其功能往往保守的.为了探索SsMYBs的功能,对这158个成员进行了GO注释.GO注释可以预测一个功能未知基因可能执行的分子功能(Molecular Function)、可能处于的细胞组分(Cellular Component)以及可能参与的生物学过程(Biological Process)[44].
簸箕柳MYB成员的GO注释中分子功能结果显示(图6),大部分成员被预测为起转录调控(46)、细胞分化(23)、气孔运动调节(7)等功能.对于细胞组分,约96%(151)的成员预测为处于细胞核内.对于生物学过程,大部分成员被预测为参与DNA 结合、转录调控区域特异性DNA结合、DNA结合转录因子活性、转录协同调节活性、序列特异性DNA结合等生物学过程.

图 6 SsMYB蛋白的GO注释Fig.6 Gene ontology annotation of SsMYB proteins
3.6 SsMYBs在簸箕柳重力刺激下的差异表达分析
R2R3-MYB家族成员参与植物对各种非生物和生物胁迫的防御和响应[18-20],并在调节植物对包括吲哚乙酸[21]、脱落酸[21, 22]、生长素[50]等植物激素线索的反应中发挥重要了作用.重力是调节植物生长和发育的一种普遍输入,各种植物谱系和器官已经进化不同的机制来调节相对于重力的生长方向[51].在被子植物中,重力刺激的反应木被称为张力木,形成于茎的上侧,且产生张力,将茎向上拉.张力木是通过维管形成层中细胞分裂速率的增加而产生的,其特征是导水导管元件数量减少,并且含有凝胶细胞壁层(G层)的特殊张力木纤维被认为是张力产生的核心[52].

图 7 SsMYB基因在簸箕柳重力刺激下的差异表达分析Fig.7 Expression analysis of SsMYB gene under gravistimulation of S.suchowensis
为了探究SsMYBs在簸箕柳应对重力刺激时可能发挥的作用,对正常茎和重力刺激下茎的张力木进行差异表达分析,选取logFC>1或<-1和padj<0.01的MYB基因为差异表达基因.共筛选出20个差异表达R2R3-MYB基因(图7),上调基因有11个,下调基因9个.其中,SsMYB103为显著下调基因,其在拟南芥中的同源基因(AtMYB15)为拟南芥激活木质素生物合成基因所必需的调节因子[53].G层的特征是低木质素、高纤维素[52].推测SsMYB103参与了G层的形成进而推动了簸箕柳茎的背地性响应.
4 讨 论
MYB家族是植物最大的转录因子家族之一,参与了植物的多种重要生物学过程,如初级和次级代谢、发育过程、生物和非生物胁迫反应、细胞和器官形态发生以及细胞周期控制[3, 54].MYB转录因子的进化和功能一直是研究的热点.MYB转录因子已在一些植物物种中被鉴定与分析,如拟南芥、水稻、马铃薯和茶树[55-58],但对于簸箕柳MYB家族的了解却很少.
本研究在簸箕柳中一共鉴定出158个R2R3-MYB转录因子家族成员.对这158个成员先后进行了保守结构域、系统进化、基因结构、基序组成、亚细胞定位、理化性质、染色体定位、基因复制事件以及GO注释等分析.
多序列比对后,发现SsMYBs结构域平均约含有105个氨基酸残基,其中,R2重复序列含有3个色氨酸残基来形成疏水核心,R3重复序列中,其疏水核心中第一个色氨酸残基被苯丙氨酸取代,第二个和第三个色氨酸残基保守性很强.相比之下,SsMYBs结构域以外的区域在长度和氨基酸组成方面的保守性较差,与之前其他植物该家族结构域的研究一致.随后进行系统进化分析,除SsMYB101未能很好地成簇外,其余157个家族成员一共被分为了23个亚组.在同一亚组中的基因通常具有相似的内含子-外显子结构和相似的基序组成.值得注意的是,某些motif只存在于特定的亚组,这些亚组特异性motif可能有助于其功能分化.簸箕柳染色体组装还不足够完善,这可能是导致7个SsMYBs无法定位到染色体上的原因,其余151个基因则在19条染色体上不均匀、无规律地分布.片段重复和串联复制是导致植物基因家族扩展的两个主要原因.在SsMYBs中存在两组串联复制事件,和83个片段重复对,这些结果表明,片段重复和串联复制在簸箕柳MYB基因家族的扩展中起主要作用.Ka/Ks比值用于估计中性突变、纯化选择和有益突变之间的平衡.计算了串联和片段复制基因对的Ka/Ks比值.结果显示,所有Ka/Ks比值均小于1,表明簸箕柳MYB复制成员在进化过程中可能经历了纯化选择压力.采用Blast2GO[44]对SsMYBs进行GO注释.大多数SsMYBs被预测位于细胞核内,约占96%.SsMYBs被预测参与簸箕柳很多的生物学过程,其中46位成员被预测为参与转录调控过程、23位成员被预测为参与细胞分化过程等.大多数SsMYBs被预测起DNA绑定分子功能.除此之外,本研究对簸箕柳茎抗重力刺激应答相关的SsMYB进行了分析.共鉴定出20个差异表达基因.其中,SsMYB103为显著下调基因,为拟南芥AtMYB15的同源基因,可能通过参与了G层的形成进而推动了簸箕柳茎的背地性响应.
综上所述,本研究对簸箕柳R2R3-MYB转录因子家族进行了全基因组水平的鉴定与分析,为簸箕柳R2R3-MYB基因功能研究和家族特性提供了全面系统的信息,并为改善簸箕柳的生长调节和抗倒伏性能提供了有价值的信息.
