基于计算机模拟技术分析去氢枞酸作为PI3K/AKT/mTOR信号通路抑制剂的潜力
2021-05-11李翠萍陈乃源李昕宇卢国栋
李翠萍,陈乃源,李昕宇,卢国栋
1广西医科大学口腔医学院;2广西医科大学公共卫生学院;3广西医科大学基础医学院;4广西高校高发疾病预防与控制研究重点实验室,南宁 530021
PI3K/AKT/mTOR信号通路对细胞的增殖、分化和凋亡等过程起着重要作用,在许多肿瘤细胞中存在过度激活的现象,并被发现可促进肿瘤细胞耐药性的产生[1,2]。而抑制该信号通路的激活能够促进肿瘤细胞凋亡,也能恢复肿瘤细胞对药物的敏感性[3]。因此,开发小分子PI3K/AKT/mTOR信号通路抑制剂成为抗肿瘤药物的研究热点之一[4]。去氢枞酸(DHA)是一种重要的三环二萜类天然树脂酸,也是传统中药松香的成分之一,主要由松香经歧化反应后分离获得。其性质稳定且具有与甾类分子类似的结构,目前已被广泛用于荧光试剂和药物中间体的合成与开发。去氢枞酸及许多它的衍生物表现出良好的抗肿瘤活性[5-7]。近期报道显示,去氢枞酸的1H苯并[d]咪唑类衍生物对PI3Kα有抑制效果,能够下调磷酸化AKT的表达[8]。我们的研究也发现,部分B环改性的去氢枞酸衍生物能够降低磷酸化的PI3K、AKT、mTOR表达水平,同样也能降低其下游效应器S6和4EBP1的磷酸化水平[9]。虽然不同去氢枞酸衍生物对PI3K/AKT/mTOR信号通路蛋白表现出了一定的抑制作用,但去氢枞酸本身是否具有相应的抑制能力,目前尚不清楚,有待进一步的研究。
大多数化合物通过与蛋白分子的结合产生效应作用,其结合模式和结合能力可通过蛋白晶体解析和蛋白荧光猝灭等手段来验证,但实验周期较长,成本较高。分子对接技术是一种利用计算机进行理论计算预测化合物与受体蛋白结合情况的手段,可根据对接结合能的大小判断化合物与蛋白的结合能力,筛选出潜在的蛋白调节剂;也可通过所得的对接构像和其他的对接评分进行初步的机理研究[10-12]。此外,了解化合物在人体的吸收、分布、代谢、排泄和毒性等类药性和药代动力学特性可以判断其是否可作为临床治疗的候选化合物。为了降低研究成本,计算机辅助程序已逐渐被用于类药性和药代动力学的初步研究[13,14]。
因此,本研究利用分子对接技术预测去氢枞酸与该通路关键蛋白的结合能力和结合方式,并用蛋白免疫印迹法验证去氢枞酸的实际抑制情况,进一步利用网络服务器进行类药性与药代动力学的模拟,以初步了解去氢枞酸作为PI3K/AKT/mTOR信号通路抑制剂的潜力,为进一步开发去氢枞酸提供理论依据。
1 材料与方法
1.1 分子对接
1.1.1 蛋白数据获取
本研究选取人癌细胞中过度激活的Ⅰ类PI3K催化亚基p110的四个亚型α、β、γ、δ[15],AKT的3种亚型AKT1、AKT2、AKT3、以及mTOR[16]进行研究。蛋白质晶体结构主要从RCSB Protein Data Bank(PDB)数据库(http://www.rcsb.org/)中获取,蛋白文件ID分别为:PI3Kα(4ZOP)、PI3Kβ(4J6I)、PI3Kγ(3L54)、PI3Kδ(4GB9)、AKT1(3MVH)、AKT2(3D0E)和mTOR(4JT6),配体均为ATP-竞争抑制剂。AKT3结构的PDB文件通过Phyre2网络服务器预测得到[17]。
1.1.2 配体文件的准备
蛋白文件中的配体分子经PyMOL Molecular Graphics System(Version 2.0 Schrödinger,LLC)提取后保留为“.pdb”格式备用。去氢枞酸(图1)和前期研究中的去氢枞酸基PI3K/AKT/mTOR信号通路抑制分子(含吡啶-3-甲基胺结构的去氢枞酸衍生物,DBDA)[9]由ChemOffice 2019绘制成3D格式,经MM-2能量优化后保存为“.pdb”格式备用。所有“.pdb”格式文件的分子,经AutoDock 4.2.6软件(AutoDock)自动选择可扭转键后保存为PDBQT文件备用。
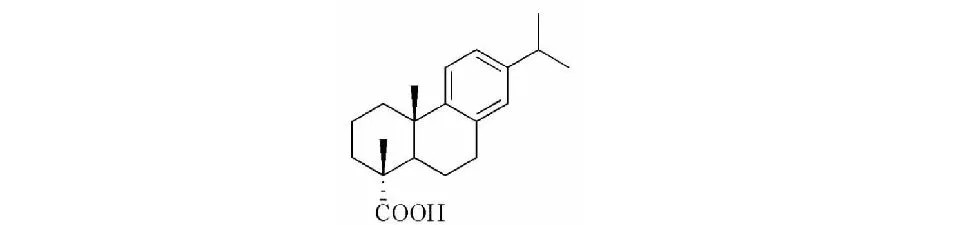
图1 去氢枞酸的结构
1.1.3 蛋白文件的准备
利用PyMOL Molecular Graphics System删除蛋白文件中的配体、水和金属等小分子,删除相似性极高的重复蛋白链,以保留A链为主。利用AutoDock对蛋白进行“加氢”、自动计算“Gasteiger charges”、设置原子类型为“Assign AD4 type”后保存为PDBQT文件备用。
1.1.4 对接的执行
“GridBox”的“grid map”设置为60×60×60以涵盖ATP位点(中心分别为:PI3Kα,x=-2.4、y=12.3、z=-17.3;PI3Kβ,x=19.0、y=60.0、z=22.0;PI3Kγ,x=22.0、y=15.0、z=22.0;PI3Kδ,x=17.0、y=-5.0、z=21.0;ATK1,x=19.0、y=-3.0、z=28.0;AKT2,x=23.0、y=-18.0、z=7.0;AKT3,x=3.0、y=15.0、z=-42.0;mTOR,x=52.0、y=1.0、z=-47.0)。对接程序使用默认参数进行,选取结合能最低的构像作为预测结果进行分析。当各蛋白原配体重新对接的构像均方根偏差(root mean square deviation,RMSD或refRMS)差小于2Å时,则认为此对接方法有较好的准确性[18]。
1.1.5 对接结果分析
配体-蛋白相互作用的3D构像使用PyMOL制作,配体-蛋白相互作用的2D构像使用LigPlot+2.1制作。去氢枞酸与原配体重叠的相互作用残基用红圈指示。相互作用残基是否参与配体-蛋白的结合通过可接触表面积(accessible surface area,ASA)的损失(ΔASA)来衡量,配体与残基的相互作用越紧密ΔASA越大,一般ΔASA大于10Å2可认为该相互作用参与了配体-蛋白的结合[19]。各残基的ASA通过Accessible Surface Area and Accessibility Calculation for Protein(ver.1.2)(http://cib.cf.ocha.ac.jp/bitool/ASA/)系统进行计算。各残基的ΔASA=ASA对接前﹣ASA对接后。
1.2 蛋白免疫印迹测试
取对数生长期的SCC9细胞,向细胞培养皿中加入含有不同浓度药物(0、13和26 μM的去氢枞酸)的培养基,置37 ℃、5%CO2培养后,用冷PBS洗,进一步用0.25%胰蛋白酶消化细胞并收集,加入200 μL裂解液(196 μL裂解液加2 μL PMSF和2 μL蛋白磷酸酶抑制剂混合物)(Solarbio)裂解,置于冰上研碎至匀浆后,4 ℃放置30 min,用BCA试剂盒检测(Beyotime)蛋白浓度,用SDS-PAGE凝胶电泳(Beyotime)对蛋白质进行电泳并转膜。在室温下用5%脱脂乳封闭膜1 h,并在4 ℃下与一抗孵育过夜。次日洗膜后,在室温下用相应的二抗孵育1 h,最后用化学发光试剂盒显影(Merck,德国)。使用全能成像系统显示蛋白条带(ChemiDoc MP,Bio-Rad,USA)。一抗为:PI3K p110、PI3K p85、p-AKT、AKT、mTOR、p-mTOR、p-4EBP1、p-S6,GAPDH作为内参(Cell Signaling Technology,Boston,USA)。
1.3 类药性与药代动力学预测
利用网络服务器“pkCSM-pharmacokinetics”(http://biosig.unimelb.edu.au/pkcsm/)对去氢枞酸的类药性(利宾斯基五法则)和药代动力学(吸收、分布、代谢、排泄和毒性)进行预测[19]。该预测方法是将已知药代动力学特性的分子进行图形建模,转化成原子间距离参数的识别标志,再用这些识别标志和分子的特性建立药代动力学的回归与分类模型。待测分子也转换成识别标志后代入模型并进行预测。
2 结果
2.1 对接结合能
对接结果显示(表1),原配体重新对接的RMSD值均小于2 Å,最大值为0.48 Å,说明对接程序的预测结果能够很好的重复原配体在蛋白中的构像,对接方法较为可靠。DHA与通路蛋白的最低对接结合能均为负值,最大值为-6.16 kcal/mol,说明DHA可能对这些蛋白的ATP位点均有一定的结合能力。虽然最低对接结合能均高于作为抑制剂的原配体,但与对该通路有抑制作用的DBDA接近。这预示着去氢枞酸可能对该通路也可以表现出抑制能力。
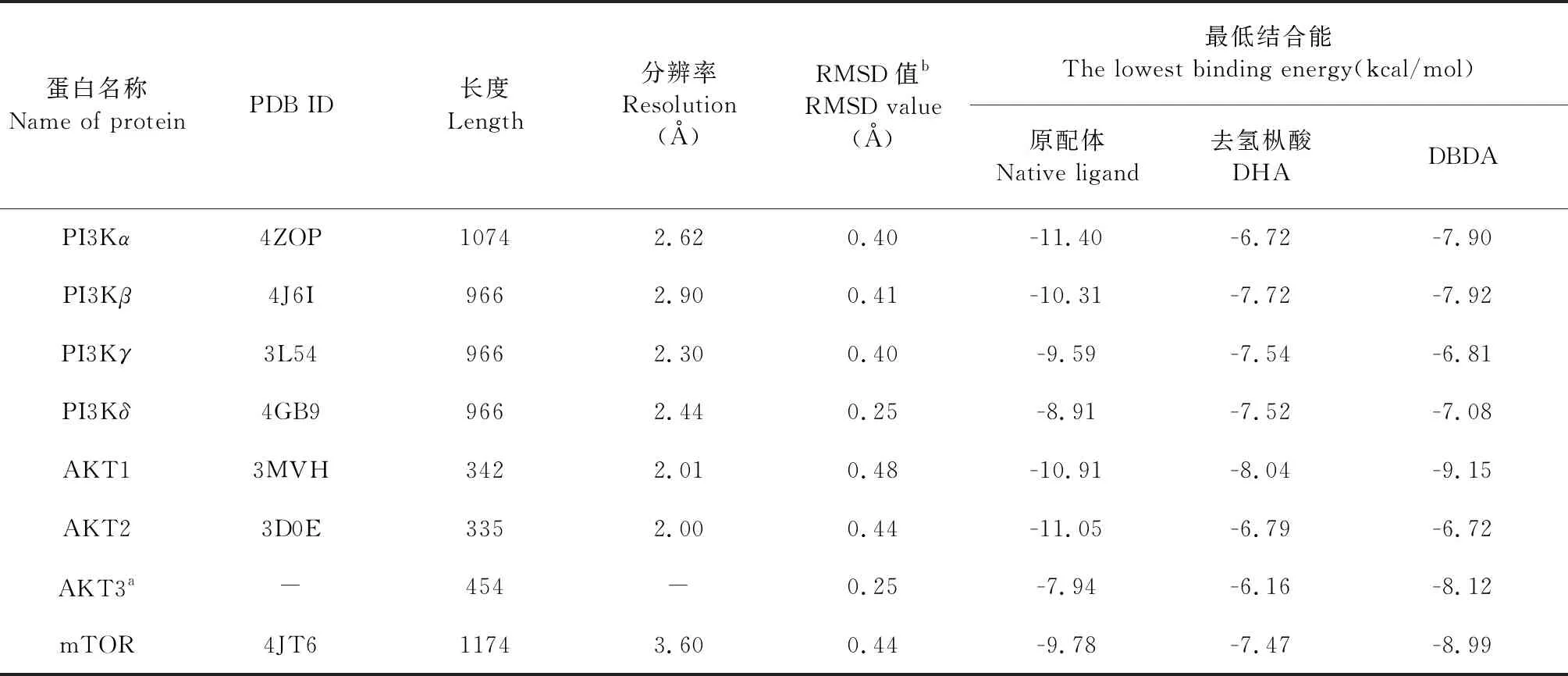
表1 PDB文件信息和最低对接结合能
2.2 去氢枞酸与各PI3K蛋白的对接结果
对PI3Kα的分子对接结果显示(图2),去氢枞酸在PI3Kα的ATP结合位点共与12个残基发生非键相互作用,其中9个残基Val851、Trp780、Met922、Met772、Ile932、Ile848、Ser854、Gln859和Arg770对配体-蛋白的结合起着重要作用(ΔASA>10 Å2),残基Trp780的ΔASA(38.99 Å2)最大,可视为关键残基(表2)。有10个残基与PI3Kα原配体的相互作用残基重叠,这说明去氢枞酸和原配体对PI3Kα的ATP结合位点有着较为相似的结合方式。

图2 去氢枞酸与PI3Kα对接的构像
对PI3Kβ的分子对接结果显示(图3),去氢枞酸在PI3Kβ的ATP结合位点与13个残基发生相互作用,其中氢键相互作用残基2个(Ala885和Lys890)、非键相互作用残基11个。有9个残基Val882、Trp812、Ala885H、Met953、Ile963、Ile831、Lys890、Ile879和Asp964对配体-蛋白的结合起着重要作用(ΔASA>10 Å2),残基Ile963的ΔASA (30.642 Å2)最大,可视为关键残基(表2)。此外,有10个残基与PI3Kβ原配体的相互作用残基重叠,这说明去氢枞酸和原配体对PI3Kβ的ATP结合位点有着较为相似的结合方式。
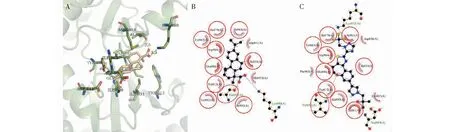
图3 去氢枞酸与PI3Kβ对接的构像
对PI3Kγ的分子对接预测显示(图4),去氢枞酸在PI3Kγ的ATP结合位点共与12个残基发生相互作用,其中与氢键相互作用残基1个(Lys890)、非键相互作用残基11个。有8个残基Met804、Asp964、Met953、Ile963、Ile831、Ser806、Ile879和Lys890对配体-蛋白的结合起着重要作用(ΔASA>10 Å2),残基Met804的ΔASA(49.215 Å2)最大,可视为关键残基(表2)。有9个残基与PI3Kγ原配体的相互作用残基重叠,这说明去氢枞酸和原配体对PI3Kγ的ATP结合位点有着较为相似的结合方式。
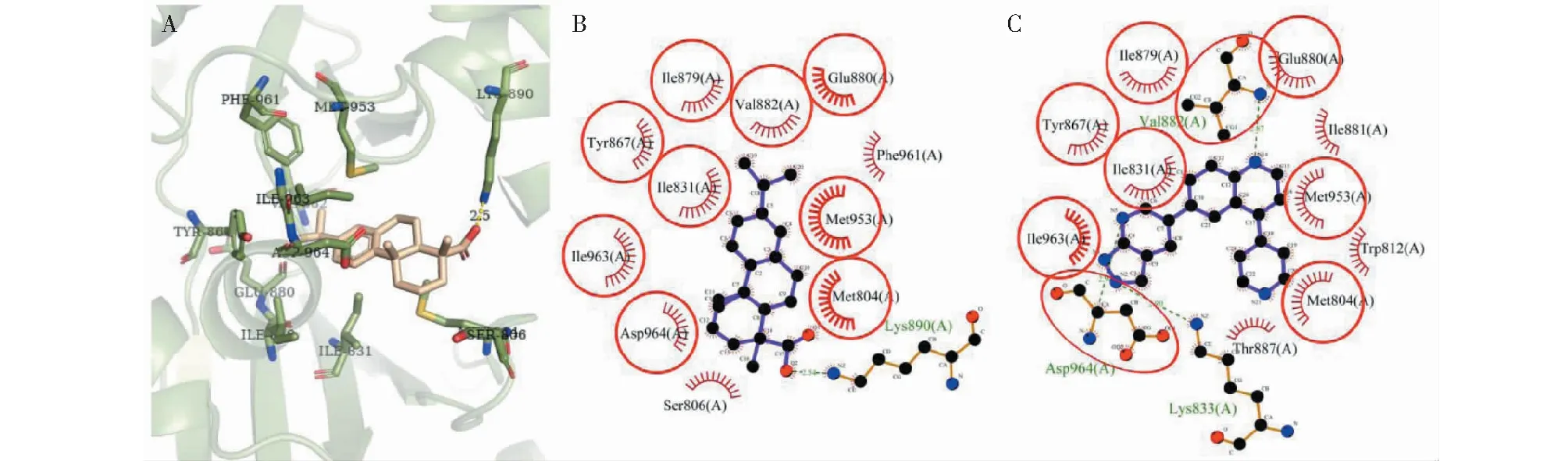
图4 去氢枞酸与PI3Kγ对接的构像
对PI3Kδ的分子对接结果显示(图5),去氢枞酸在PI3Kδ的ATP结合位点与13个残基发生相互作用,其中氢键相互作用残基1个(Lys890)、非键相互作用残基12个。有7个残基Met953、Lys890、Ile963、Met804、Ile879、Ile831和Ser806对配体-蛋白的结合起着重要作用(ΔASA>10 Å2),残基Lys890的ΔASA(43.805 Å2)最大,可视为关键残基(表2)。有11个残基与PI3Kδ原配体的相互作用残基重叠,这说明去氢枞酸和原配体对PI3Kδ的ATP结合位点有着较为相似的结合方式。
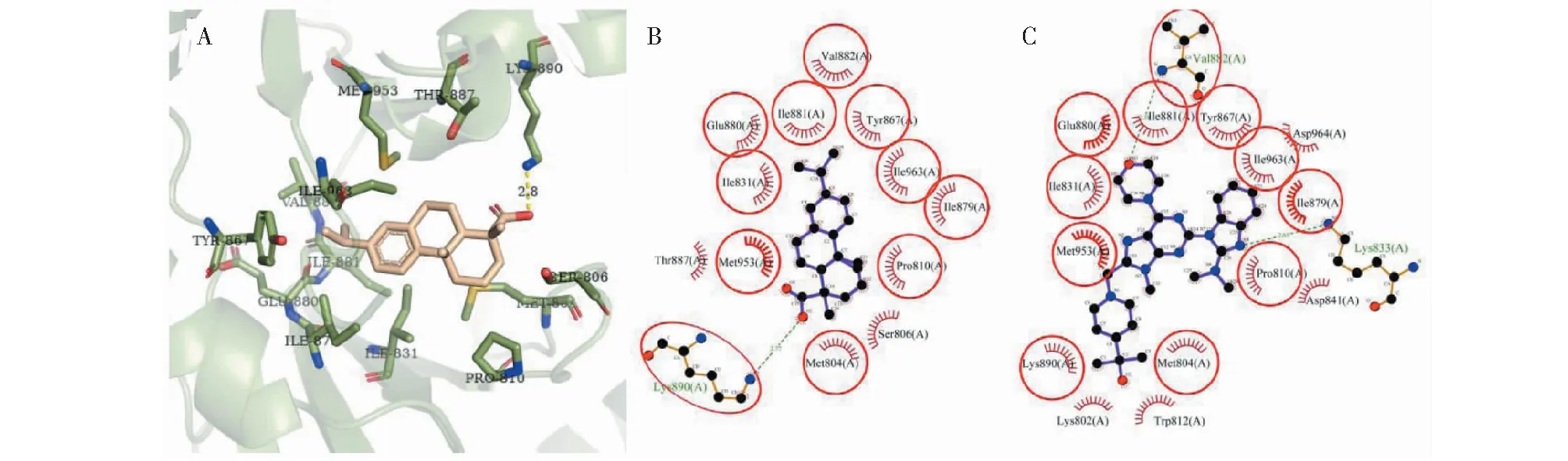
图5 去氢枞酸与PI3Kδ对接的构像
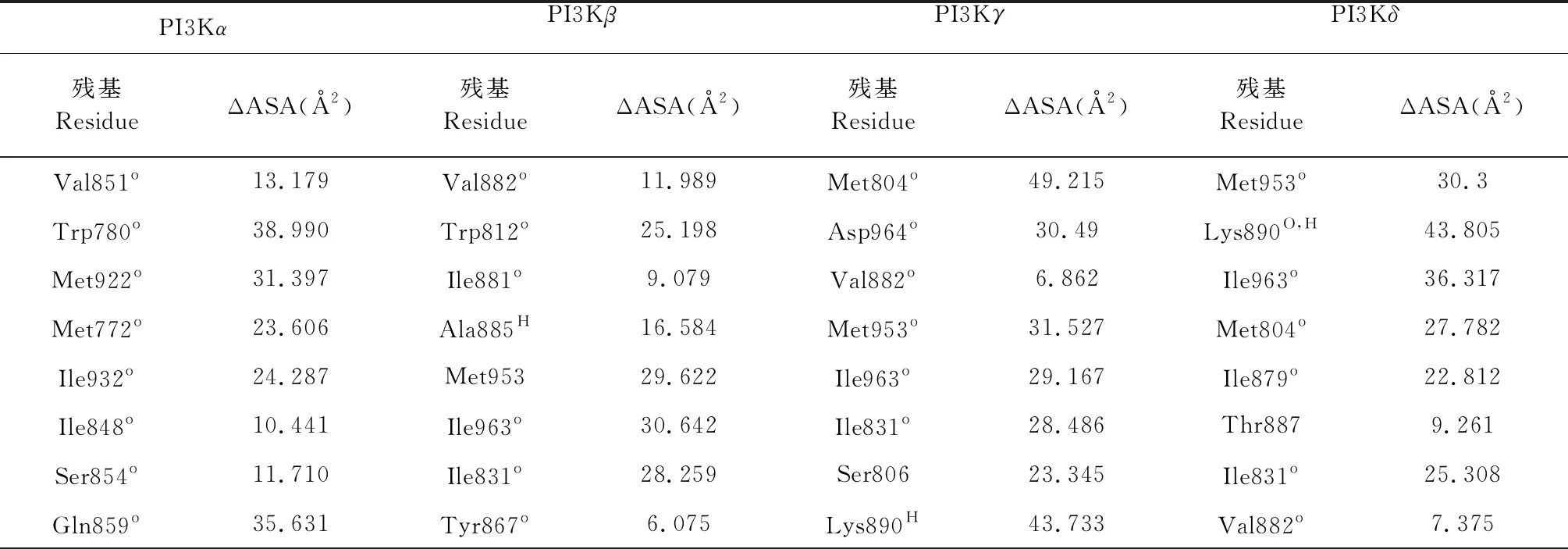
表2 去氢枞酸与PI3K相互作用残基信息

续表2(Continued Tab.2)
2.3 去氢枞酸与各AKT蛋白的对接结果
对AKT1的分子对接结果显示(图6),去氢枞酸在AKT1的ATP结合位点与15个残基发生相互作用,其中氢键相互作用残基2个(Thr211和Ala230)、非键相互作用残基13个。有10个残基Glu234、Asp292、Val164、Leu156、Arg4 (B)、Met281、Met227、Gly157、Ala177和Thr291对配体-蛋白的结合起着重要作用(ΔASA>10Å2),残基Val164的ΔASA (40.176Å2)最大,可视为关键残基(表3)。有12个残基与AKT1原配体的相互作用残基重叠,这说明去氢枞酸和原配体对AKT1的ATP结合位点有着较为相似的结合方式。

图6 去氢枞酸与AKT1对接的构像
对AKT2的分子对接结果显示(图7),去氢枞酸在AKT2的ATP结合位点与13个残基发生非键相互作用,其中8个残基Lys181、Asp293、Val166、Glu236、Met229、Gly159、Thr292和Met282对配体-蛋白的结合起着重要作用(ΔASA>10Å2),残基Val166的ΔASA (30.756Å2)最大,可视为关键残基(表3)。有11个残基与AKT2原配体的相互作用残基重叠,这说明去氢枞酸和原配体对AKT2的ATP结合位点有着较为相似的结合方式。
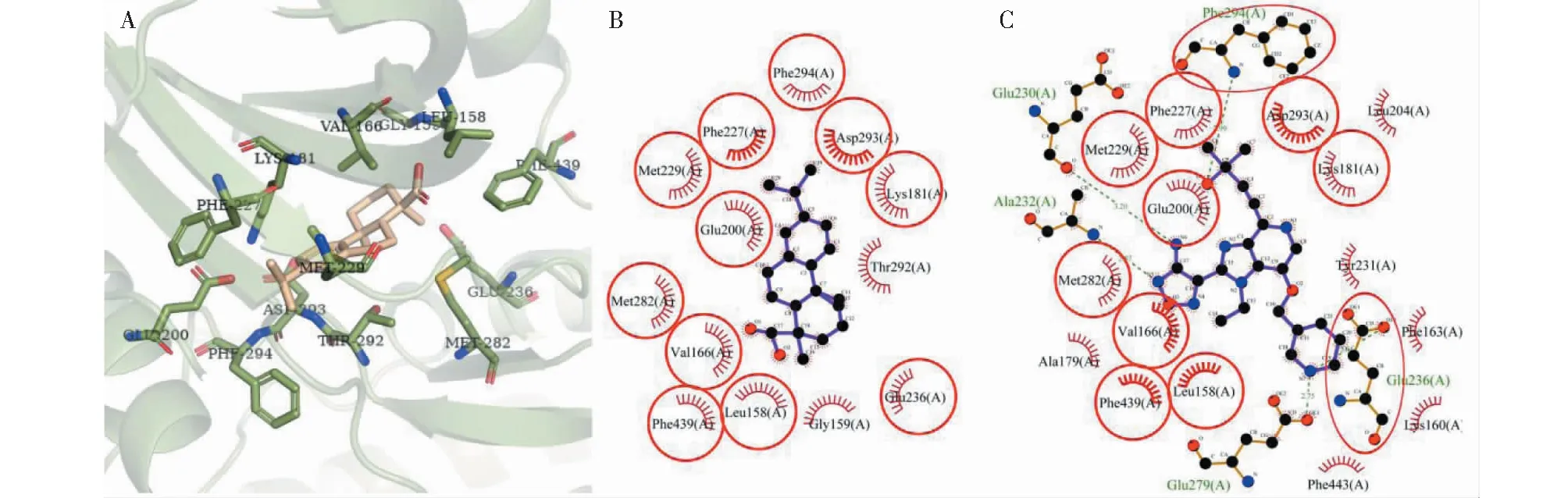
图7 去氢枞酸与AKT2对接的构像
对AKT3的分子对接结果显示(图8),去氢枞酸在AKT3的ATP结合位点与14个残基发生非键相互作用,其中9个残基Leu154、Val228、Val162、Met278、Gly155、Met225、Asp289、Lys177和Gly157对配体-蛋白的结合起着重要作用(ΔASA>10Å2),残基Met278的ΔASA(41.586Å2)最大,可视为关键残基(表3)。近半数的残基与AKT1原配体的相互作用残基重叠,说明去氢枞酸与AKT1配体在一定程度上对AKT3的ATP结合位点有着相似的结合方式。
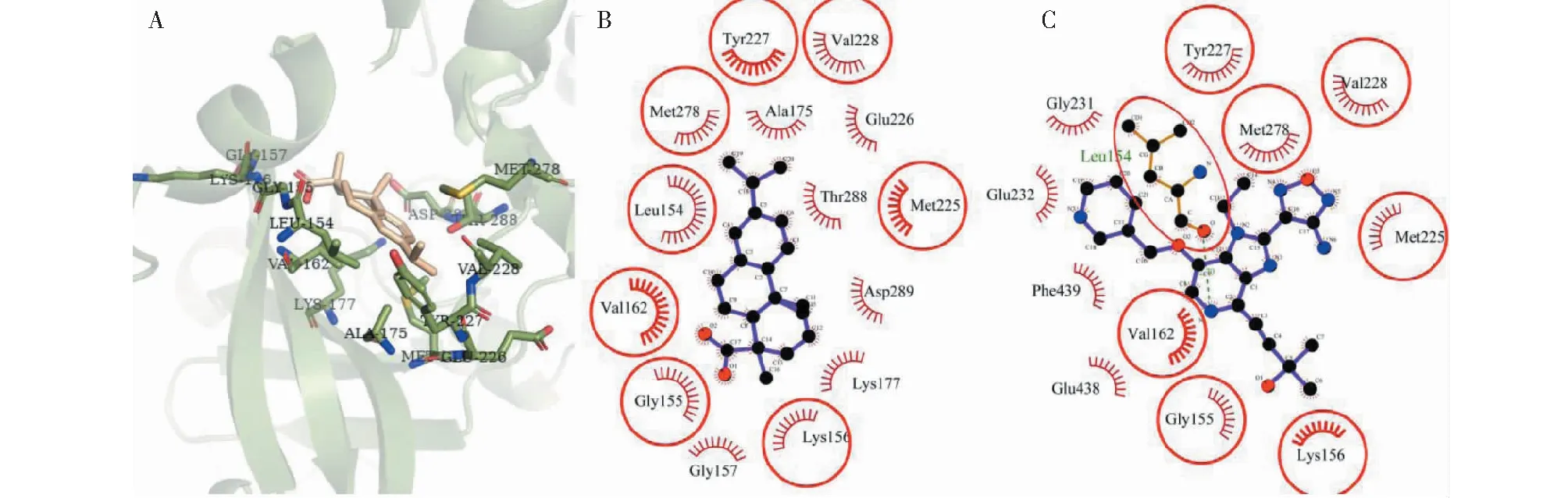
图8 去氢枞酸与AKT3对接的构像

表3 去氢枞酸与AKT相互作用残基信息
2.4 去氢枞酸与mTOR蛋白的对接结果
对mTOR的分子对接结果显示(图9),去氢枞酸在mTOR的ATP结合位点与13个残基发生相互作用,其中氢键相互作用残基1个(Val2240)、非键相互作用残基12个。有9个残基Trp2239、Ile2356、Ile2237、Met2345、Leu2185、Cys2243、Thr2245、Ala2248和Asp2244对配体-蛋白的结合起着重要作用(ΔASA>10 Å2),残基Trp2239的ΔASA(50.969 Å2)最大,可视为关键残基(表4)。近半数的残基与mTOR原配体的相互作用残基重叠,这说明去氢枞酸与原配体可能在一定程度上对mTOR的ATP结合位点有着相似的结合方式。
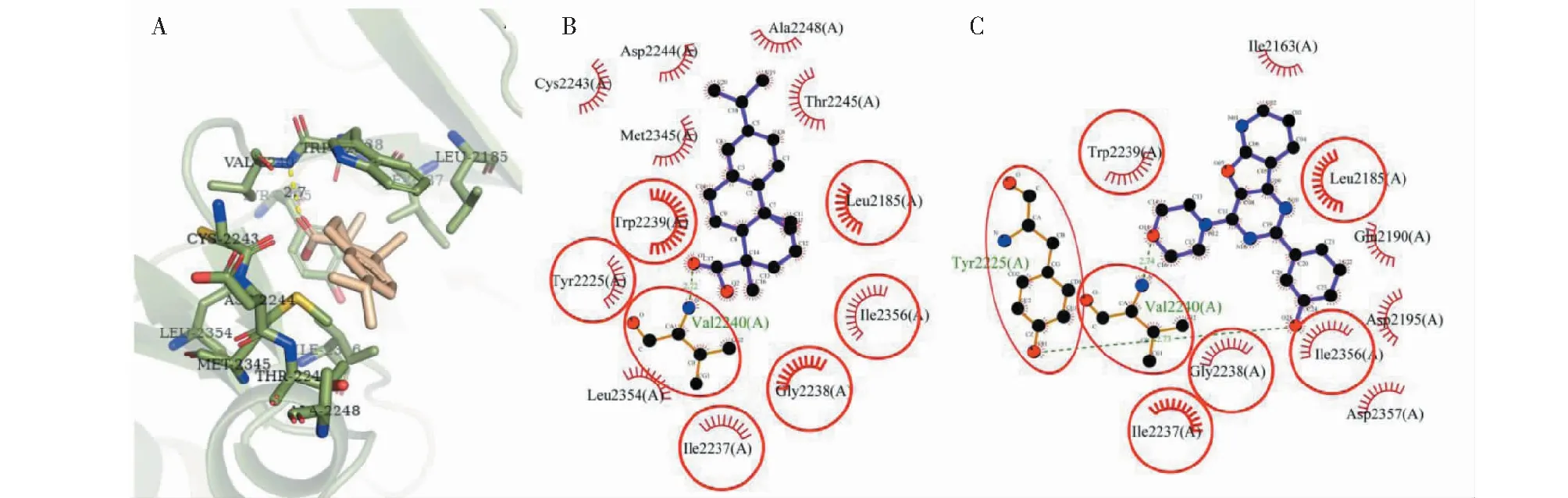
图9 去氢枞酸与mTOR对接的构像

表4 去氢枞酸与mTOR相互作用残基信息
2.5 蛋白免疫印迹结果
蛋白免疫印迹结果显示(图10),在去氢枞酸的作用下,SCC9的PI3K催化亚基p110的表达略有下调,调控亚基p85的表达明显下调;PI3K下游蛋白AKT和mTOR的磷酸化明显被抑制,但其总蛋白的表达基本不受影响;通路下游的效应蛋白4EBP1的磷酸化略受抑制,S6的磷酸化明显减少。这说明PI3K/AKT/mTOR信号通路受到去氢枞酸的抑制。
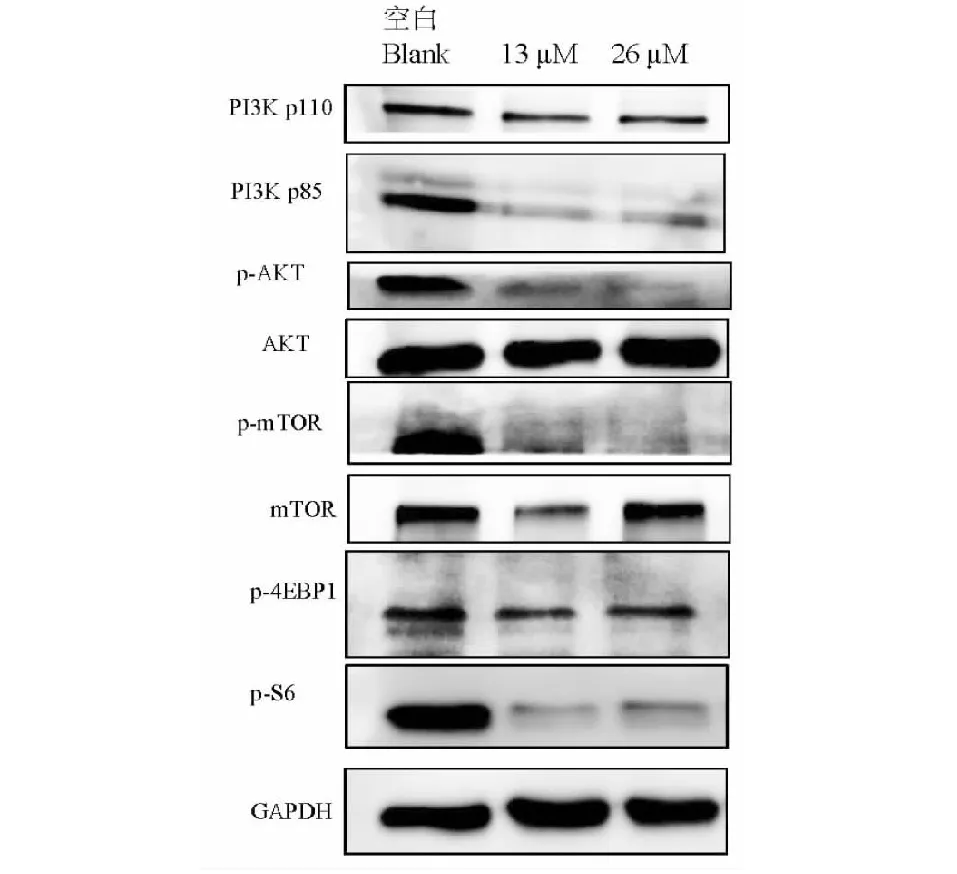
图10 去氢枞酸对蛋白表达的影响
2.6 类药性与药代动力学预测结果
类药性预测结果经“五倍率法则(Lipinski rule of five)”比较(表5):去氢枞酸的分子量、油水分配系数(LogP)、可旋转键(rotatable bonds)、氢键受体(acceptors)、氢键供体(donors)和总极化表面积(surface area)均在可接受值域内。
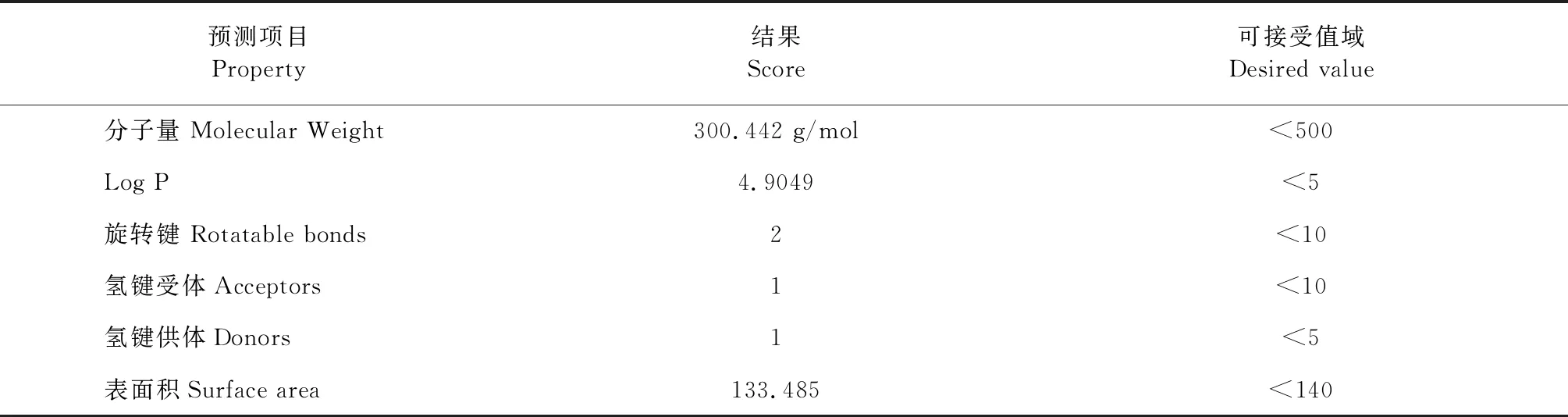
表5 去氢枞酸类药性预测数据
药代动力学预测结果显示(表6):去氢枞酸有较好的脂溶性和口服作用,容易通过肠道吸收,且不作为P-糖蛋白的底物;较易分布于血液,易穿透血脑屏障、穿透中枢神经;对机体的代谢能力没有影响;可以被正常排泄;不致突变,毒性较小。总的来说去氢枞酸通过了大多数的预测,成为安全药物的可能性较大,可考虑作为治疗使用的候选化合物[19]。
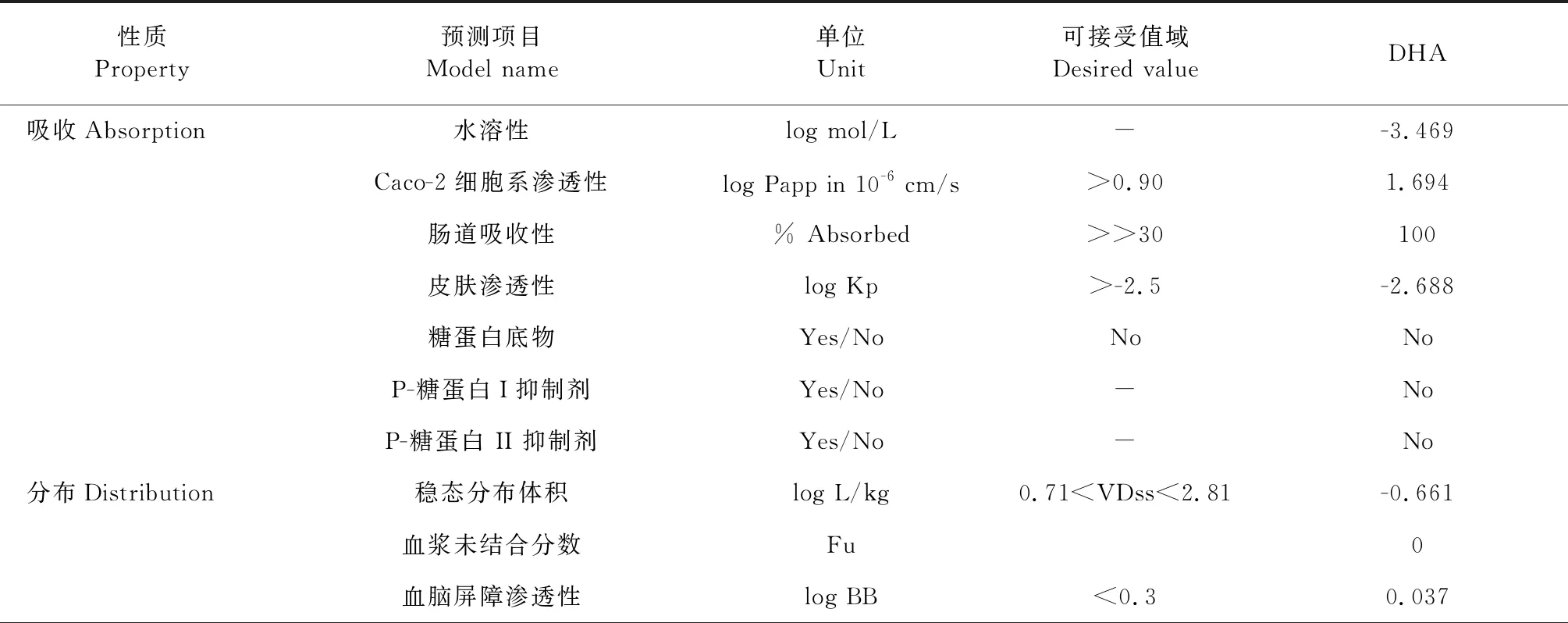
表6 去氢枞酸药代动力学预测数据
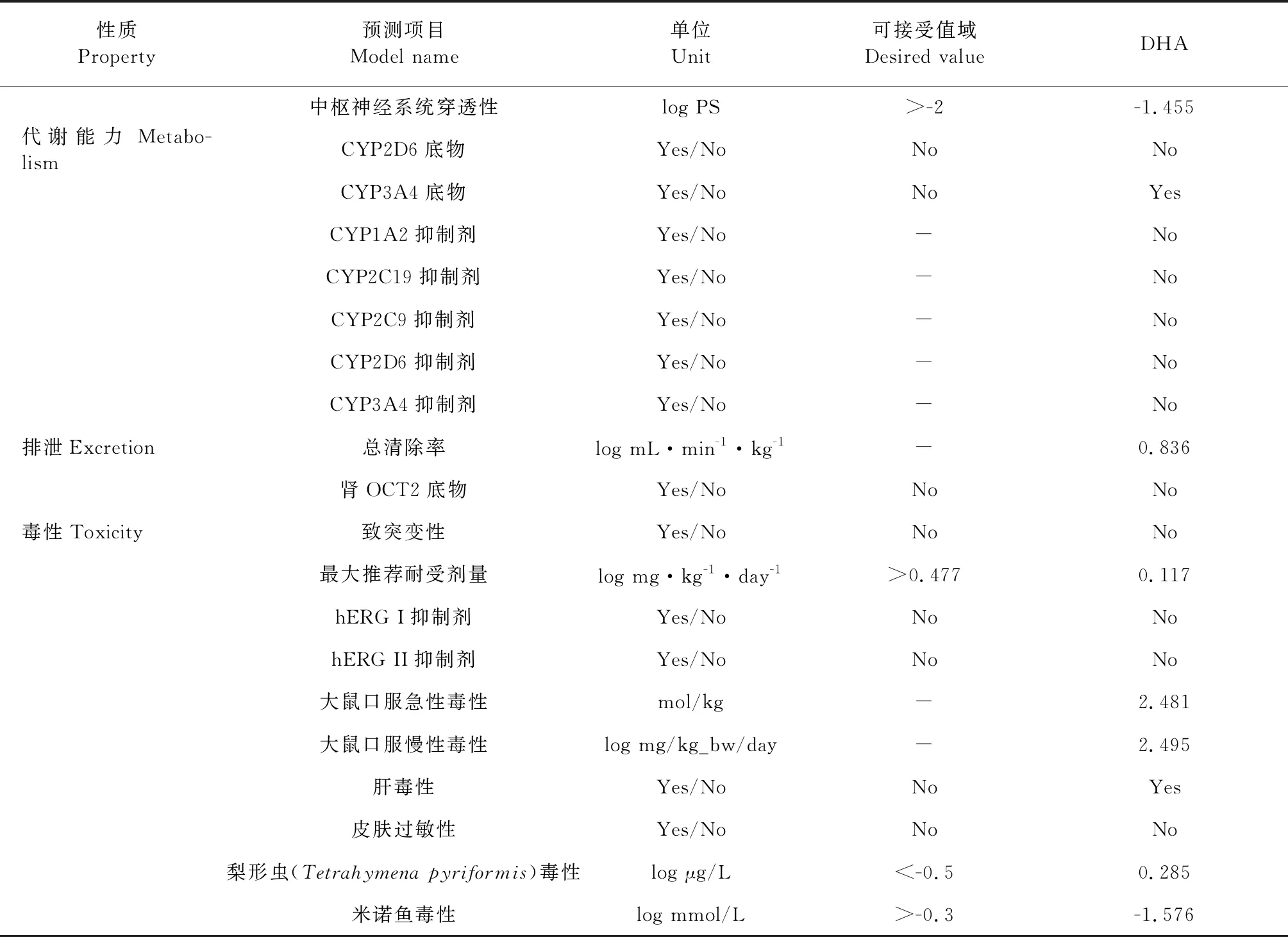
续表6(Continued Tab.6)
3 讨论
PI3K/AKT/mTOR信号通路对细胞的增殖、分化和凋亡等过程起着重要作用,也是癌症治疗的靶点之一。PI3K主要由催化亚基p110和调控亚基p85构成,经磷酸化激活后,可将其底物3,4-二磷酸磷脂酰肌醇(PIP2)转化成3,4,5-三磷酸磷脂酰肌醇(PIP3)。PIP3能够与AKT结合后通过磷酸肌醇依赖性蛋白激酶1(PDK1)促使AKT磷酸化。经磷酸化激活的AKT能够直接或间接的使mTOR磷酸化,从而进一步促进下游效应蛋白4EBP1和S6的磷酸化以调控细胞内生化活动[20,21]。
本研究基于近年来去氢枞酸抗癌衍生物不断被开发,部分衍生物被报道对PI3K/AKT/mTOR信号通路的部分关键蛋白有抑制作用[8,9,22]。为了研究作为原料的去氢枞酸对PI3K/AKT/mTOR信号通路的影响,为去氢枞酸抗癌衍生物的开发提供依据。利用分子对接技术预测了去氢枞酸对通路关键蛋白PI3K,AKT和mTOR的ATP结合位点的结合能力和结合方式,利用蛋白免疫印迹法检验去氢枞酸对通路的抑制效果,通过初步的类药性和药代动力学模拟预测去氢枞酸成为口服药物的潜力。
分子对接结果表明,去氢枞酸对各关键蛋白的ATP结合位点均有一定的结合能力,与AKT3的结合最弱,最低结合能为-6.16 kcal/mol,与AKT1结合最强,最低结合能为-8.04 kcal/mol。去氢枞酸对通路蛋白的结合能力弱于作为高效抑制剂的原配体,与我们已合成得到的去氢枞酸基PI3K/AKT/mTOR信号通路抑制分子DBDA相近。在ATP结合位点中,去氢枞酸与蛋白的相互作用残基多为含疏水性残基。除了PI3Kδ的Lys890,其他对配体-蛋白结合起最重要作用的关键残基也均为疏水性残基。说明去氢枞酸可能通过与ATP结合位点的腺嘌呤区结合从而产生抑制作用[23]。各蛋白与去氢枞酸的相互作用残基跟原配体的有着大量的重叠,这表明去氢枞酸可能是以和原配体相近的方式与ATP展开竞争以抑制蛋白作用。此外,没有氢键存在的构像,如去氢枞酸与PI3Kα、AKT2和AKT3,它们的结合能均高于其他存在氢键的构像,这表明氢键对去氢枞酸与蛋白的稳定结合发挥着重要作用。在去氢枞酸结合的构像中,氢键全部在羧基位置产生。但是,异丙基和苯环附近存在多个在原配体中为氢键相互作用的重叠残基。这一特征提示,在设计新的去氢枞酸基PI3K/AKT/mTOR信号通路抑制剂时,可加强羧基位置的亲水能力,并在异丙基和苯环位置引入亲水基团,可能可以提高新化合物与蛋白的结合能力。
蛋白免疫印迹实验显示,SCC9细胞经去氢枞酸处理后,PI3K调控亚基p85的表达明显降低。p85含量的减少可能使PI3K的生成受阻,并成为减少PI3K下游蛋白AKT磷酸化表达的因素之一。AKT和mTOR的总蛋白在不同浓度的去氢枞酸作用下与空白组相比没有明显的变化,但其磷酸化蛋白表达均明显减少,说明AKT和mTOR的磷酸化进程受到抑制。处于mTOR下游的4EBP1的磷酸化表达下降较少,可能是因为4EBP1还可经MEK/Erk信号通路激活的原因[24]。而受mTOR调控的另一效应蛋白S6K1下游的S6磷酸化表达明显减少,可能是因为mTOR的磷酸化受阻,导致S6K1激活减少,从而减少了S6的磷酸化反应。总的来说在SCC9细胞内,PI3K/AKT/mTOR信号通路的表达受到了去氢枞酸的抑制,这与分子对接中预测的结果一致。
类药性和药代动力学预测中,去氢枞酸的五倍率法则(Lipinski rule of five)分析显示,其分子量、LogP、旋转键、氢键受体、氢键供体的值均在该经验性规则要求的范围内[25]。药代动力学预测对去氢枞酸在体内的吸收、分布、代谢、排泄和毒性等性能进行模拟,去氢枞酸通过了大部分的测试,说明它可能可以较好的在体内发挥药物作用[19]。
总之,本研究通过分子对接预测、蛋白免疫印迹实验、类药性检验和药代动力学预测,发现去氢枞酸本身可能成为一种候选的治疗药剂,用于抑制PI3K/AKT/mTOR信号通路,以达到缓解肿瘤细胞耐药和抗肿瘤的目的。也可以进一步对去氢枞酸的结构进行修饰,以开发出更有效的去氢枞酸基PI3K/AKT/mTOR信号通路抑制剂。
