罗红霉素未知杂质的分离鉴定与抗菌活性研究
2022-06-16孟祥燕冯传威樊伟明周炜祥王建红刘小睿沈永淼
孟祥燕 冯传威 樊伟明 周炜祥 王建红 刘小睿 沈永淼,*
(1 浙江震元制药有限公司,绍兴 312000;2 浙江理工大学理学院化学系,浙江省高分子材料表界面科学重点实验室,杭州 310018)
红霉素(erythromycin, EA)最初是由Lilly公司分离得到的首个大环内酯类抗生素药物,是近半个世纪以来使用最广泛的高效抗多数革兰阳性菌和部分革兰阴性菌的抗生素。主要由内酯环、德糖胺和克拉定糖组成[1-2]。红霉素在酸性条件下不稳定,口服后易在胃酸作用下生成无抗菌活性的螺缩酮衍生物,生物利用率低[3]。为提高其稳定性,人们对涉及反应位点的C-6,C-9,C-11及C-12等进行结构改造,抑制环合降解,合成了一系列半合成的红霉素衍生物。
红霉素可分为红霉素A、B、C、D、E、F,其中红霉素E的制备仅在1973年的美国专利(US3714142)中有报导[4]。红霉素ABCDE均有一定的抗菌活性,但活性差异较大,红霉素A是红霉素的主要活性成分。红霉素B在抗菌谱和抗菌活性上与A相似,但毒性为A的两倍;红霉素C不是活性物质,因此B和C除了在发酵中需控制含量,后续处理时也应除去,以提高活性物质A的含量。红霉素E(图1)由于含量低,相关研究较少,抗菌活性仅为A的13%[5],一般作为非活性物质除去[6-7]。
前期研究发现,红霉素E和羟胺反应衍生化之后的产物—红霉素E肟的抗菌活性也远不如红霉素A肟[8]。罗红霉素杂质G'这个杂质以往是由红霉素E经一系列反应形成的(图2),均直接归类到罗红霉素的未知杂质组分中,目前仅有一篇文献通过质谱推测了其可能的结构[9],没有明确的杂质研究及活性研究,以往均作为非活性物质通过结晶工艺除去,国家标准中要求其含量<0.5%,额外的纯化工艺增加了罗红霉素的成本。
本文通过制备液相色谱对罗红霉素杂质G'进行有效分离提纯,通过结构表征确定了该杂质为红霉素E的衍生化产物,同时用前期确定结构的杂质肟进行进一步衍生化反应,同样能得到罗红霉素杂质G',进一步佐证了结构的正确性。抗菌活性实验结果显示,该杂质与罗红霉素的抗菌活性相当,说明罗红霉素杂质G'有成为新型抗菌药物的潜力,同时,对罗红霉素的生产工艺来说,如果经试验证实罗红霉素杂质G'的毒性与罗红霉素相当,在后续的生产中可降低罗红霉素杂质G'的质控要求。
1 仪器与材料
高效液相色谱仪(GTXH-3000;安捷伦科技有限公司),色谱柱(C18,5 μm,4.6 mm×250 mm),核磁共振谱仪(AVANCEⅢ-400M;瑞士布鲁克拜厄斯宾有限公司),质谱仪(Thermo LCQ Fleet spectrometer;美国热电公司),硅胶(烟台江友硅胶开发有限公司),二氯甲烷、氨水(萨恩化学技术有限公司),甲醇(杭州高晶精细化工有限公司),9-(E)-红霉素A肟粗品及罗红霉素粗品均来自于浙江震元制药有限公司。
2 实验方法
2.1 罗红霉素杂质G'的分离
2.1.1 从9-(E)-红霉素A肟杂质中分离
在前期的研究中已经从9-(E)-红霉素A肟的粗品中分离出9-(E)-红霉素E肟[8]。本实验将已经纯化的3 g 9-(E)-红霉素E肟粗品溶解在12 mL N,N-二甲基甲酰胺(DMF)溶剂中,升温至全溶,然后用冰水降温度至10℃以下,加入0.8 mL甲醇钠甲醇溶液,搅拌,继续降温,在恒压滴液漏斗中,加入约10 mL DMF和1-甲氧基-2-氯甲氧基乙烷0.52 g,当料液降温至0℃以下,缓慢滴加,加完后搅拌,反应结束后用醋酸调pH至6.5~7.5,将料液移至浓缩罐浓缩,控制内温小于65℃,至基本浓缩干后,加入3.0~5.0倍甲醇溶解,加入适量活性炭,保持温度35℃~45℃脱色30 min以上,过滤,料液移至结晶罐中,用液碱调pH至10.5~11.5,再加入甲醇体积1.5~2.5倍的工艺用水进行结晶,将固体离心甩干,真空干燥得产量为2.2 g,经液相色谱及质谱检测,罗红霉素杂质G'的产率为70%。
2.1.2 从罗红霉素中分离
取2.05 g罗红霉素样品溶解于适量CH2Cl2通过制备液相色谱进行第一次分离,富集到纯度为50%含量的罗红霉素杂质G',然后再取500 mg 1次分离后的杂质G'(50%左右含量)进行2次分离,所用的制备层析条件如下:固定相:硅胶(200~300目,厂家:烟台江友硅胶开发有限公司);洗脱剂:体积比CH2Cl2:CH3OH:NH4OH=50:1:1;检测波长:210 nm;流速:30 mL/min。
2.2 罗红霉素杂质G'的结构确证
2.2.1 由前体肟的结构间接确证
在前期发表的文章[8]中已经报道了中间体杂质肟的结构。从该杂质肟出发,通过醚化反应,得到了罗红霉素杂质G',由此间接确证了杂质G'的结构。
2.2.2 谱图表征直接确证
从罗红霉素粗产品中直接分离所得到的杂质G',通过IR、1H-NMR、13C-NMR、HRMS、2D-NMR等表征手段进行结构确证。
2.3 抗菌活性研究
用微量稀释法测定所分离的杂质对革兰阳性菌(金黄色葡萄球菌、肺炎球菌、草绿色链球菌以及白念珠菌)及革兰阴性菌(大肠埃希菌)的最小抑菌浓度。本实验采用微量液体二倍稀释法确定化合物对各种细菌的最小抑菌浓度(MIC)[10]。
3 分析与讨论
3.1 杂质的结构分析与鉴定
3.1.1 核磁共振波谱分析
将分离得到的罗红霉素杂质G'用CDCl3溶解,进行核磁共振波谱分析,其1H-NMR和13C-NMR谱图见图3~4,数据及归属见表1~2。
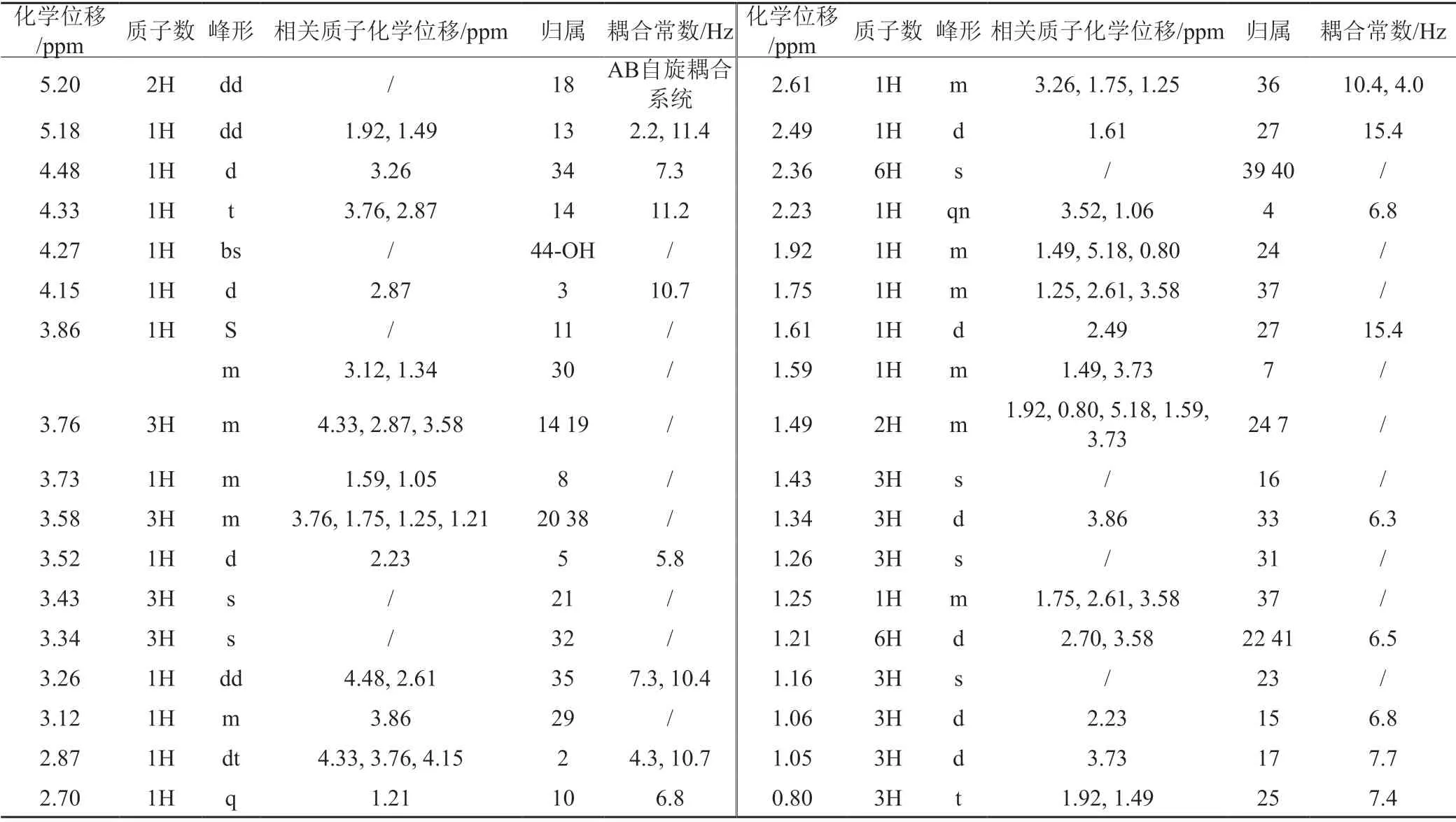
表1 罗红霉素G' 1H NMR数据及归属Tab.1 The 1H-NMR date and assignment of the roxithromycin impurity G'
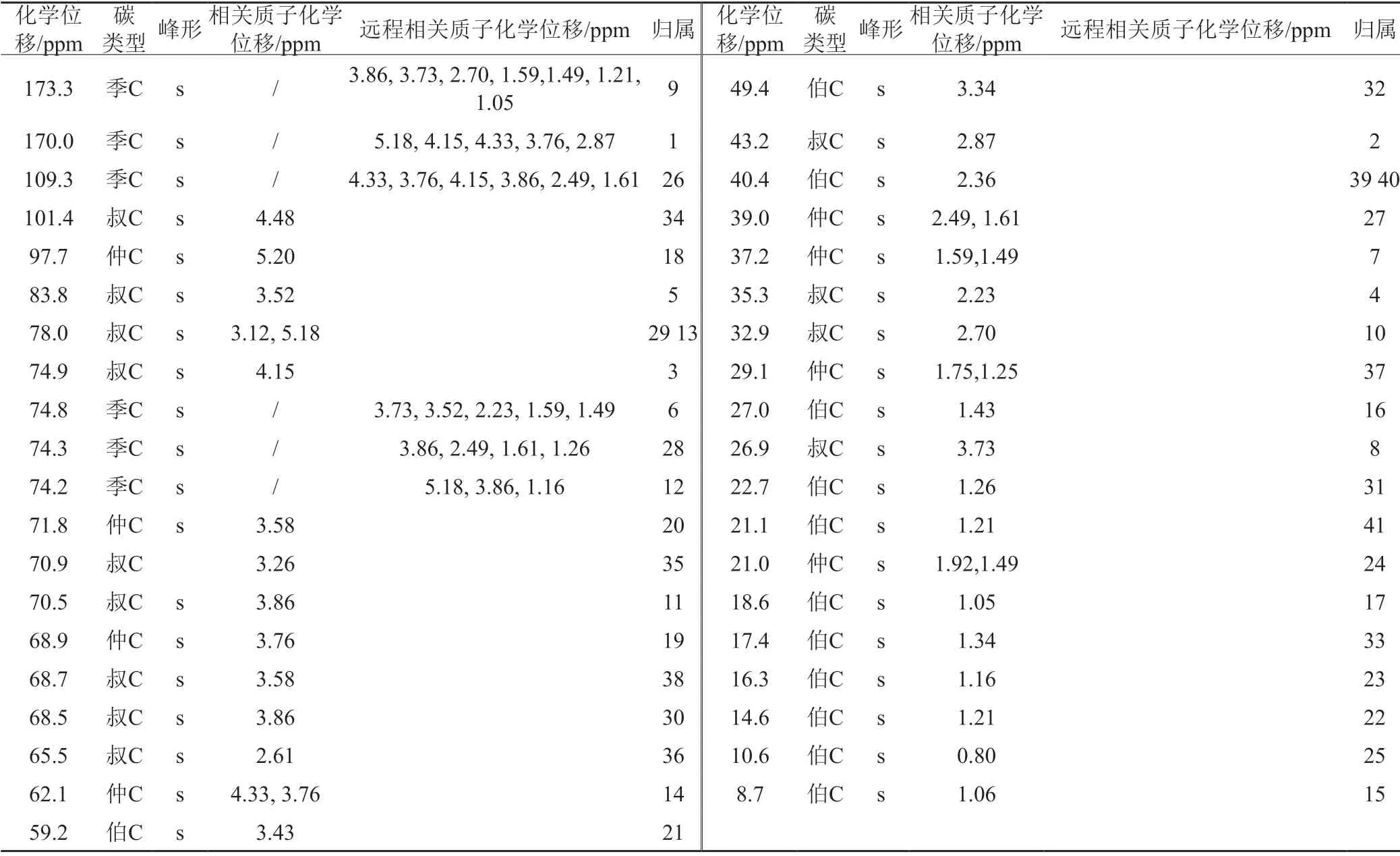
表2 罗红霉素G' 13C NMR数据及归属Tab.2 The 13C-NMR dateandassignment of theroxithromycin impurity G'
3.1.2 结构鉴定
红外吸收光谱在3433 cm-1:O-H伸缩振动;1106 cm-1:C-O伸缩振动;1106 cm-1:O-H面外弯曲振动,1632、1592 cm-1:C=N伸缩振动;2833 cm-1:-C-H伸缩振动;1358 cm-1:C-H弯曲振动。上述峰位显示该杂质中有-OH、C=N、饱和烷烃结构,与罗红霉素G' 结构相符。同时,13C-NMR δ173.3 ppm的季碳峰也验证存在C=N;13C-NMR δ170.0 ppm的季碳峰表明存在脂羰基结构。除此之外1H-NMR、13C-NMR峰的化学位移与结构一致,表明与罗红霉素G'分子结构吻合。HRMS(ESI-TOF)m/z:[M+H]+计算C41H75N2O16的分子量为851.5117,谱图测得分子量为851.5105,与 G'分子式相符,同时在紫外光谱近紫外区没有强吸收峰,表明该分子结构中无共轭体系。
该结构中的2、3、4、5、6、8、10、11、12、13、28、39、30、34、35、36和38位是手性碳,其中2位碳氢和3位碳氢发生偶合,偶合常数为10.7 Hz,推断出2位H和3位H处于反式,都位于直立a健。其它手性碳构型依据罗红霉素G'反应前体——罗红霉素肟的单晶结构推断确定。从上述推断可以得出罗红霉素G'的结构式(图5)。
3.2 杂质的抗菌活性研究
以罗红霉素为参照物,对罗红霉素杂质G'进行了抗菌活性研究,结果(表3)显示,罗红霉素杂质G'的抗菌活性与罗红霉素相当,对肺炎球菌、草绿色链球菌的抗菌活性较好,对白念珠菌未显示出抗菌作用,因此该杂质有成为新型抗菌药物的潜力。
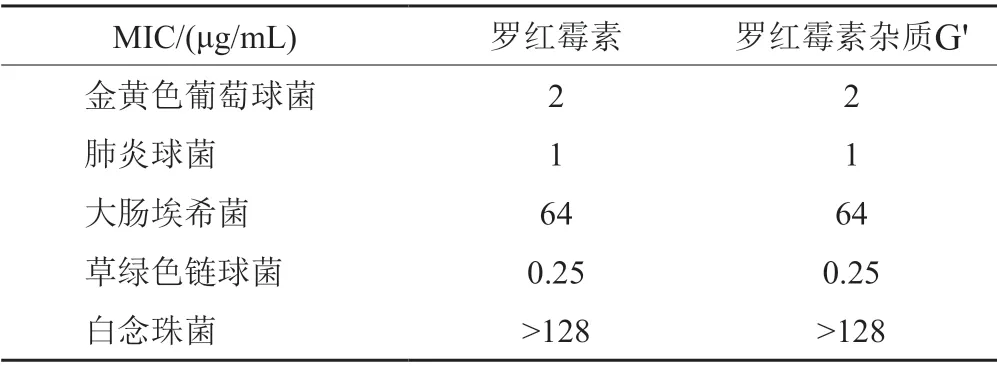
表3 罗红霉素杂质G'的最低抑菌浓度Tab.3 The minimal inhibitory concentrations (MIC) of roxithromycin impurity G'
4 结论
本文通过液相色谱法来分离罗红霉素中的一个主要未知杂质罗红霉素杂质G',然后通过1HNMR,13C-NMR、IR、HRMS等来确定未知杂质的化学结构为红霉素E衍生化产物,最后测试了该杂质对金黄色葡萄球菌,肺炎球菌,大肠埃希菌,草绿色链球菌及白念珠菌的抗菌活性,与罗红霉素比较发现两者的抗菌活性相当。因此,认为通过对红霉素E的结构进行适当的修饰有望合成一类新型抗菌药物。且若两者毒性和体内抗菌活性也相当,则在后续生产过程中可降低罗红霉素G'的质控要求,大大降低了生产成本。
