四种不同生境条件下外来植物长芒苋入侵对土壤细菌群落组成和多样性的影响
2022-06-09石福臣张梅
石福臣,张梅
(南开大学 生命科学学院,天津 300071)
0 引言
长芒苋(Amaranthus palmeri)属苋科(Ama⁃ranthaceae)、苋属植物,原产美洲,长芒苋的一些特性使其成为全球性恶性杂草并难以防除,例如光合C4途径,其光合速率是大豆和棉花的3~4倍[1-2];极高的繁殖能力和生长速率,自然状态下每株长芒苋可产生60万粒种子,株高可达2 m~3 m;极强的极端环境的耐受能力和遗传变异能力[3]。此外,长芒苋对多种除草剂产生抗药性更使其成为世界头等恶性杂草[4]。
长芒苋于1985年首次发现于中国北京市丰台区,经过近30年的潜伏传播后,近几年在京津冀地区呈现爆发性入侵与扩散趋势,迅速入侵至农田、果园等经济作物生长区,与经济作物进行养分竞争,抢夺水分、光照资源,有的甚至比经济作物(玉米、果树)还高,这对当地的农业生态系统和生物多样性安全造成巨大的威胁和破坏。国内对于长芒苋的研究较少,主要包括:李慧琪等预测了长芒苋在中国的潜在分布区,长芒苋在中国的潜在分布集中在华北平原地区[5];曹晶晶等研究表明:表型变异(如高纬度种群开花时间提前)可提高长芒苋环境适应性进化,拓展其在中国的适生性分布区,增强其向原产地适应环境梯度之外地区入侵和扩散蔓延的潜力[6];本研究组前期进行了关于长芒苋表型可塑性的研究[7]。
外来植物的入侵机制十分复杂,包括增强竞争能力、化感作用、根际理化性质改变以及微生物区系的变化等[8-10]。近年来,从植物-土壤-微生物群落组成研究外来植物的入侵机制已成为新的研究思路和重要的发展趋势。紫茎泽兰入侵会改变土壤微生物的多样性和群落结构,引起有益功能菌聚集和土壤酶活性的改变,形成有利于紫茎泽兰入侵扩张的微生态环境[11];Smooth brome入侵增加了土壤细菌的丰富度和均匀度,最重要的作用是选择性抑制优势菌种,使较少见的细菌相对丰度增加[12];Robinia pseudoacacia入侵主要改变了土壤细菌的丰富度,这种改变主要是由放线菌门、芽单胞菌门和硝化螺旋菌门等细菌门类所驱动[13];薇甘菊(Mikania micrantha)通过改变土壤微生物群落丰度,从而调节土壤碳的循环利用实现入侵[14];加拿大蓟(Cirsium arvense)通过改变土壤微生物降低一些本地植物的性能,但自身的性能并没有因此而改变,进而使加拿大蓟形成竞争优势[15]。Cheng等对于入侵植物欧洲千里光(Senecio vulgaris)根际和内生细菌群落的研究发现了许多和解磷(Brevundimonas diminu⁃ta)、固氮(Rhizobium leguminosarum)、耐受极端环 境(Exiguobacterium sibiricum)相 关 的 细菌[16]。因此,从土壤微生物角度揭示长芒苋的入侵机制,从而抑制其进一步的扩散和蔓延,具有重要的理论和现实意义。
本研究通过野外调查取样,结合三代高通量测序技术,研究四种典型生境下有无长芒苋入侵对土壤微生物多样性和种类的影响。旨在探讨以下问题:(1)不同生境下长芒苋入侵是否会影响土壤细菌群落组成和多样性;(2)不同生境下是否存在有助于长芒苋入侵的不同类型的核心微生物。
1 材料与方法
1.1 样地概况及样品采集
本研究采样点位于天津市武清区(117°16ˊE,39°59ˊN),该地区属温带半湿润大陆性季风气候,海拔年均气温11.6℃,年降水量606 mm(图1)。该地区为长芒苋重度入侵区域,并且在多种生境下形成单优群落,是进行长芒苋土壤细菌群落多样性研究的理想取样点。其主要伴生草本植物有狗尾草(Setaria Viridis)、藜(Chenopodium album)、虎尾草(Chloris virga⁃ta)、玉米(Zea mays)等。
图1 采样点及生境设置。A,河岸生境、B,路边生境、C,荒地生境、D,农田生境Fig.1 Location of research site and four types of sampling habitats:riverbank(A),roadside(B),wasteland(C)and farmland(D)habitats
本研究选择河岸(A)、路旁(B)荒地(C)和农田(D)等4种典型生境类型,每种生境设置3个采样点,每个采样点间隔30 m~50 m。每个采样点分为长芒苋入侵区(盖度大于70%)和非入侵区,每个区域设置面积为2 m×2 m的样方,两个区域间隔3 m~5 m。每个样方土壤采集采用五点取样法,去除表面的枯枝落叶后,用土钻收集0~10 cm处的土壤,混合为一个样品。共采集4(生境)×2(土壤类型)×3(重复)=24份土壤样品。所有土壤样品放入液氮罐中迅速带回实验室,剔除可见根系和石块并混匀后过4 mm筛,在−20℃冰箱中保存暂存,通过北京百迈客公司高通量测序平台进行微生物测序。
1.2 试验方法
1.2.1 土壤DNA提取、PCR扩增和高通量测序
使用MN NucleoSpin 96 Soil试剂盒对土壤样品基因组DNA进行提取。选用引物27F(5′-AGAGTTTGATCCTGGCTCAG-3′) 和 1492R(5′-GGTTACCTTGTTACGACTT-3′)对 细 菌16S全长进行扩增。PCR扩增体系(50 μL)包括:基因组 DNA 40 ng~60 ng,2.5 μL*Vn F(5′端引物)(10 μmol·L-1),2.5 μL*Vn R(3′端引物)(10 μmol·L-1),1 μL KOD FX Neo(TOYO⁃BO),25 μL KOD FX Neo Buf(2X)。 10 μL 2 mmol∙L-1dNTP,补 ddH2O 至总体系 50 μL。反应程序为:95℃预变性5 min,30个循环包括(95℃,30;50℃,30;72℃,1 min/1 kb);72℃延伸5 min。PCR结束后,对产物进行琼脂糖凝胶电泳。PCR产物经磁珠法回收后,纯化后的产物进行Nanodrop 2000定量后,按照质量比1∶1进行混样。由北京百迈客生物科技有限公司进行后续的样品建库,上PacBio测序平台进行高通量测序,利用单分子实时测序(SMRT Cell)的方法对marker基因进行测序。
1.2.2 数据分析
对原始下机subreads进行校正得到CCS(Circular Consensus Sequencing)序 列(SMRT Link,version8.0),然后使用 lima(v1.7.0)软件,通过barcode序列识别不同样品的CCS序列并去除嵌合体(version 8.1),得到高质量的CCS序列。在相似性97%的水平上对序列进行聚类,以测序所有序列数的0.005%作为阈值过滤 OTU(Operational Taxonomic Unit)。通过Venn图呈现各样本所共有和独有OTU所占的比例,应用Mothur versionv.1.30软件对Alpha多样性指数进行分析,计算ACE、Chao1、Shan⁃non和Simpson等物种多样性指数。采用SPSS 20.0软件进行双因素方差分析,评价入侵、生境及其交互作用对细菌群落alpha多样性指数的影响。为了将不同生境的聚类可视化,基于binary-jaccard构建非度量多维尺度图(NMDS)。此外,利用线性判别分析(LDA)和LEfSe方法探究不同生境下的生物标志物。运用R软件进行各种分析图的绘制与结果输出。
2 结果与分析
2.1 测序结果
24个样品测序后通过Barcode识别后共获得180 441条CCS序列,平均产生7 518条CCS序列。利用Mothur软件进行rarefaction分析,绘制稀疏性曲线(图2)。随着序列数目增加,各处理的稀疏性曲线趋于平缓。说明扩增序列可以真实反映各个土壤样品的细菌群落结构,测序深度足够。
2.2 异质生境下入侵区和未入侵区OTU分析
Venn图可以直观的体现土壤细菌群落OTU组成的差异性及重叠情况(图3)。在河岸(A)、路边(B)、荒地(C)和农田(D)4种生境中,入侵样地分别检测到1 814、1 647、1 593和1 608个细菌OTUs,未入侵样地分别检测到2 105、1 615、1 909和1 448个细菌OTUs。其中入侵样地,A、B、C、D四种生境各自独有314、145、117和240个OTUs,而在非入侵样地分别独有 213(A)、201(B)、127(C)和 104(D)个OTUs(图 2)。
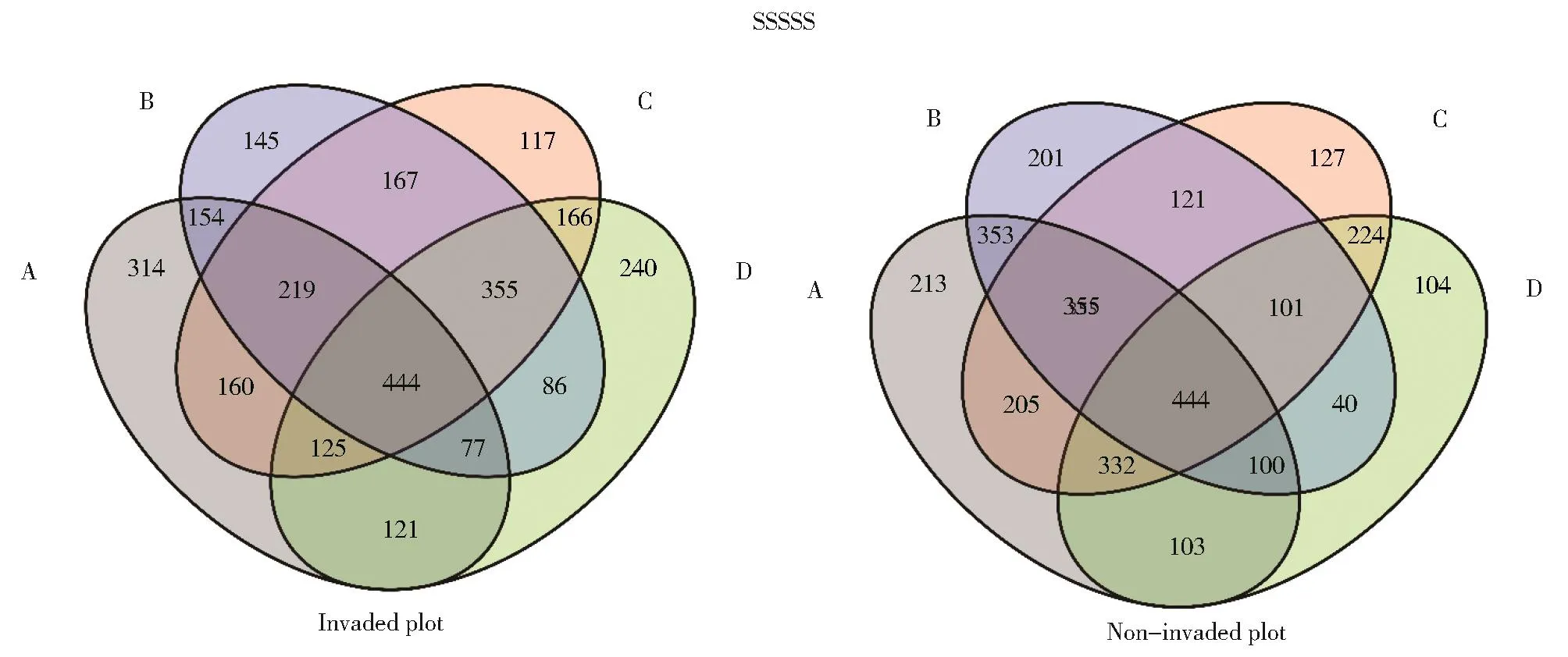
图3 四种生境有无长芒苋入侵的Veen图。A、B、C、D分别表示河岸、路边、荒地和农田生境Fig.3 Venn diagrams of shared OTUs(number of OTUs)of invaded(left)and non-invaded(right)byA.palmeri plants under four sampling habitats.Letters in the figure,A:riverbank,B:roadside C:wasteland D:farmland
2.3 土壤细菌群落Alpha多样性分析
用Chao1和ACE指数衡量物种丰度即物种数量的多少。由表1可知,河岸生境和荒地生境土壤细菌的Chao1指数和ACE指数较高,ACE指数在生境C和D间存在显著差异(P<0.05)。Shannon和Simpson指数用于衡量物种多样性,Shannon指数值越大,Simpson指数值越小,说明样品的物种多样性越高。由表1可知,荒地生境土壤细菌的Shannon指数值最高,且显著高于河岸生境(P<0.05),河岸生境土壤细菌的Simpson指数最高,且显著高于生境荒地和农田生境(P<0.05)。这表明土壤细菌群落的物种丰富度和多样性因环境的不同而存在较大差异。
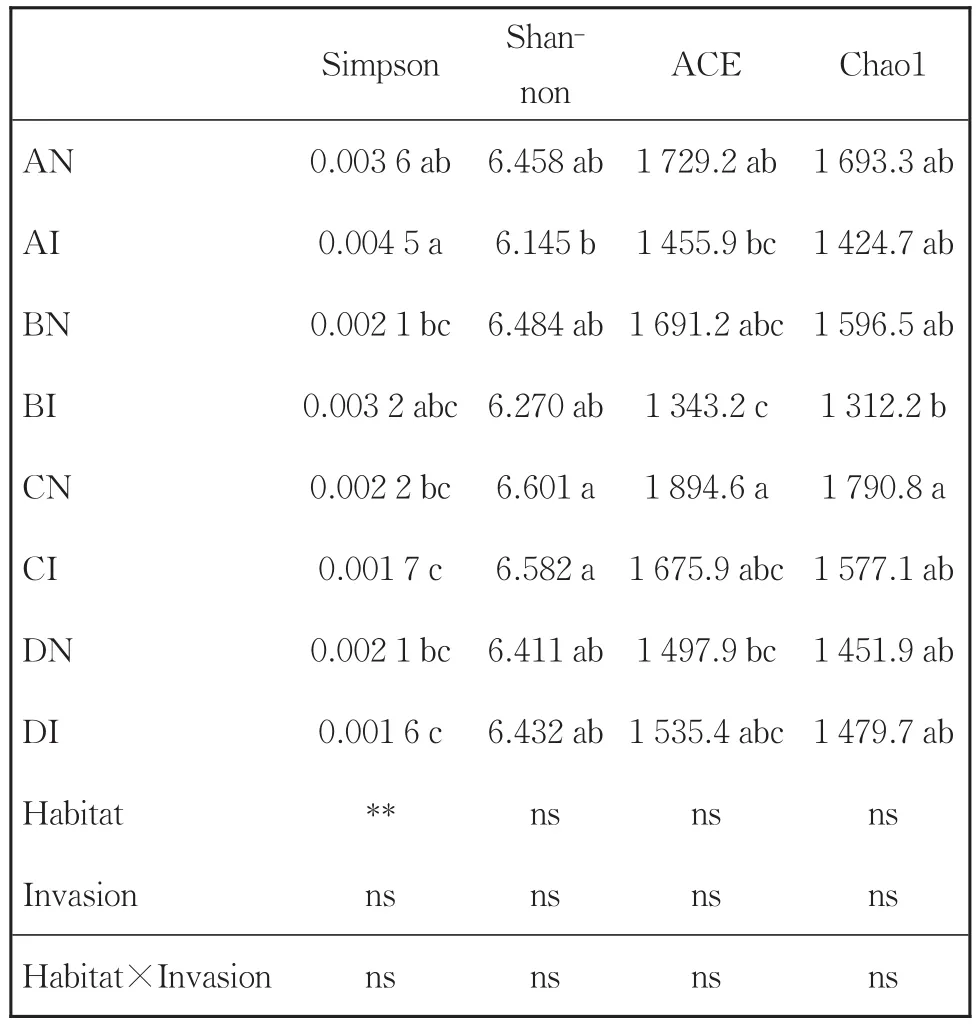
表1 异质生境下入侵和非入侵样地细菌群落的Alpha多样性Table 1 Alpha diversity of bacterial community in the invasive and non-invasive plots under four habitats
长芒苋入侵区和未入侵区土壤细菌的Chao1、ACE、Shannon、Simpson指数均不存在显著差异(P>0.05)。双因素方差分析表明:除生境对Simpson指数具有显著影响(P<0.05)外,入侵、生境及二者的相互作用均未对Alpha多样性产生显著影响(P>0.05),表明长芒苋入侵对土壤细菌群落的物种丰富度和多样性未造成较大的影响。
2.4 土壤细菌群落在门水平上的组成差异
细菌门分类水平的组成表明,4种生境中的主要优势门类为变形菌门、浮霉菌门、拟杆菌门、芽单胞菌门和酸杆菌门(图4)。变形菌门是丰度最高的类群,A、B、C、D四种生境下未入侵区的相对丰度为37.91%、29.71%、27.15%和27.42%,入侵区的相对丰度为42.75%、26.08%、30.35%和28.41%。浮霉菌门是第大二优势门类,非入侵样地的相对丰度分别为7.61%(A)、13.41%(B)、19.24%(C)、20.57%(D),入侵样地的相对丰度分别为5.38%(A)、21.79%(B)、19.78%(C)、21.34%(D)。

图4 4种生境下长芒苋入侵区和未入侵区土壤细菌群落在门水平的分类组成。图例显示了按物种数量分类的前10个物种。其他的分类为“Others”,未注释的物种分类“Unclassified”。A、B、C、D分别表示河岸、路边、荒地和农田生境Fig.4 Phylum-level taxonomic composition of the bacterial community in the soil ofA.palmeri invaded and non-invaded plots under four habitats.The legend at the top right shows the top 10 species classified by species abundance.The others are classified as "Others",and the unannotated species are classified as "Unclassified"
与非入侵区相比,河岸和荒地生境下长芒苋土壤中变形菌门的相对丰度分别增加了12.77%和11.79%,路边生境下浮霉菌门的丰度增加了62.49%,而河岸生境下浮霉菌门的丰度减少了41.45%。更重要的是,与非入侵区相比,长芒苋入侵区土壤细菌的第三大细菌门由芽单胞菌门转变为拟杆菌门。这些变化主要与河岸、荒地和农田三种生境中拟杆菌门丰度的明显增加以及芽单胞菌门的丰度明显降低有关。
2.5 长芒苋入侵四种生境下指示微生物的筛选
采用LEfSe(线性判别分析效应量)分析方法,鉴定4种生境中有显著差异的细菌类群(LDA>4.0)。从内到外的分类图代表了从门到种的分类水平(图5)。与非入侵区相比,河岸生境下长芒苋土壤细菌中的差异微生物为β-变形菌目、伯克氏菌科、Saccharimonadaceae科、黄单胞 菌 科、Flavisolibacter属、Candida⁃tus_Saccharimonas种和Flavisolibacter_sp种;荒地生境下长芒苋入侵区的核心类群为芽单胞菌纲和芽单胞菌科;同时,亚硝化单胞菌科和Phycisphaeraceae科在农田入侵生境中的相对丰度较高;路边生境下仅Sphingomonas_sediminic⁃ola种被鉴定为关键的土壤微生物类群(表2)。
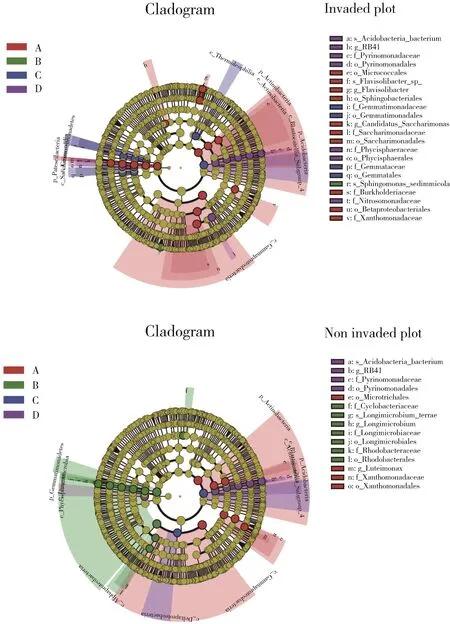
图5 采用线性判别分析(LDA)对不同生境下长芒苋入侵区(上)和无入侵区(下)的LEfSe分析。不同生境中不同类群的微生物丰度差异显著,用彩色圆点表示,圆圈表示从门到种的系统发育水平。A、B、C、D分别表示河岸、路边、荒地和农田生境Fig.5 Cladograms via linear discriminate analysis(LDA)of effect size(LEfSe)with an LDAscore higher than 4.0 ofA.palmeri invasive(top)and non-invasive(bottom)treatments under heterogeneous habitats.Significant differences in microbial abundances according to taxa among habitats are represented by colored dots.Circles represent phylogenetic levels from kingdom to genus

表2 异质生境下入侵和非入侵样地Biomarker差异分析Table 2 Variation analysis of Biomarker in the invasive and non-invasive plots under four habitats
3 讨论
大量研究证实土壤微生物群落在外来植物入侵过程中扮演了十分重要的角色[13,17-18],研究长芒苋入侵对土壤微生物多样性的影响是揭示其入侵机制的重要途径。
本研究发现,长芒苋入侵区土壤细菌OTU数量、微生物多样性指标(Chao1、ACE、Shan⁃non、Simpson)均略低于未入侵区,但均未呈现显著差异(P>0.05)(表1)。这与大多数的研究不同,但该结果与Kamutando等对于Acacia dealbata的研究有相似的结论,并且Kamutando认为微生物群落分析方法不能准确地估计微生物的丰富度,在微生物群落生态学和生物多样性研究中应慎重地应用多样性指数[19-20],因此,多种方法的结合可能会使信息获得存在一个最佳的平衡点[19,21]。
在细菌门分类水平上,变形菌门、浮霉菌门、拟杆菌门和芽单胞菌门是相对丰度较高的门类,这些菌群在其他植物入侵区也有大量报道,包括日本蓝莓[22],Acacia dealbata[20]和Cir⁃sium arvense[23]等。变形菌门是丰度最高的优势类群,我们的研究结果也进一步证实了变形菌门是陆地土壤生态系统细菌群落的优势菌群[24]。与非入侵样地相比,长芒苋入侵河岸生境通过聚集生长速度较快的细菌门类,如变形菌门,减少生长速度较慢细菌门类的累积(如酸杆菌门和浮霉菌门),形成对自身有利的生长环境,以在该生境中占据优势地位[24-26]。浮霉菌门可能在长芒苋入侵路边生境中具有重要意义,其在该生境具有极高的富集比例,这可能是因为浮霉菌门在生态系统氮和碳循环中发挥重要作用[14,27]。
LEfSe分析表明,长芒苋入侵河岸生境下的土壤优势细菌类群为伯克氏菌科和β-变形菌目。芽单胞菌纲(Gemmatimonadetes)和芽单胞菌科(Gemmatimonadaceae)在长芒苋入侵荒地生境过程中发挥重要作用,长芒苋入侵路边生境和农田生境的核心细菌类群分别为Sphin⁃gomonas_sediminicola和亚硝化单胞菌科。β-变形菌目、伯克氏菌科和亚硝化单胞菌科均隶属于变形菌门,前人研究表明这些微生物类群在生态系统化学循环过程中发挥着积极的生态作用[28-30]。例如菌类在固氮,氨氧化以及保护植物免受土壤病原体侵害等作用[31-32]。Sphin⁃gomonas_sediminicola于2013年首次从淡水沉积物中分离出来[33],Sphingomonasspp.已显示出促进植物生长的积极作用[34]。长芒苋入侵土壤中鞘氨醇单胞菌的显著增加,一方面加速土壤碳氮循环,另一方面减轻农药、除草剂等污染物对自身的毒害作用,形成对自身有利的环境,加速其入侵[35]。值得注意的是,尽管与非入侵样地相比,长芒苋入侵的荒地土壤中芽单胞菌门的丰度呈下降趋势,但芽单胞菌纲和芽单胞菌科仍是该生境的优势类群。表明这些微生物在长芒苋入侵荒地过程中发挥了重要作用。
4 结论
本文探讨了在异质性生境下,长芒苋入侵对土壤细菌群落组成及多样性的影响。结果表明:长芒苋入侵虽然对土壤细菌α多样性未产生较大的影响,但对细菌群落的组成和结构影响较大。本研究在不同生境条件下筛选出了一种到几种可作为长芒苋土壤细菌群落结构变化的指示种。未来研究可以结合土壤酶、分泌物、功能基因等方面分析,进一步探究导致微生物群落改变的内在机制。
