骨关节炎发病机制及药物调控新进展
2022-06-08韩明睿刘倩倩
韩明睿,刘倩倩,孙 洋
(南京大学生命科学学院生物技术与药学系,医药生物技术国家重点实验室,江苏 南京 210023)
骨关节炎(osteoarthritis,OA)是一种复杂的慢性疾病,是老年人致残的主要原因,其病变涉及关节软骨、软骨下骨、滑膜和关节周围肌肉的结构改变,主要表现为骨摩擦、晨僵、疼痛和关节运动障碍等[1]。随着全球人口老龄化和肥胖加剧,OA对人类的影响逐年加重。在中国,1990年约有26.1万人患OA,这一数字在2017年上升到61.2万人,其年龄标准化患病率为3.1%,其发病率女性高于男性[2]。世界卫生组织预测,我国将成为世界OA患病人数最多的国家。因此,探究OA的分子发病机制,研究治疗OA的药物一直是世界骨科领域研究的焦点问题。本文将对近年关于OA发病机制的研究及药物调控的进展进行概述。
1 参与OA发病的主要细胞
目前,关于骨关节炎发病机制的相关研究认为主要与软骨代谢稳态失衡,软骨下骨硬化和滑膜炎症密切相关,涉及巨噬细胞、T细胞、软骨细胞、骨骼干细胞、破骨细胞等多种细胞,如Fig 1所示。
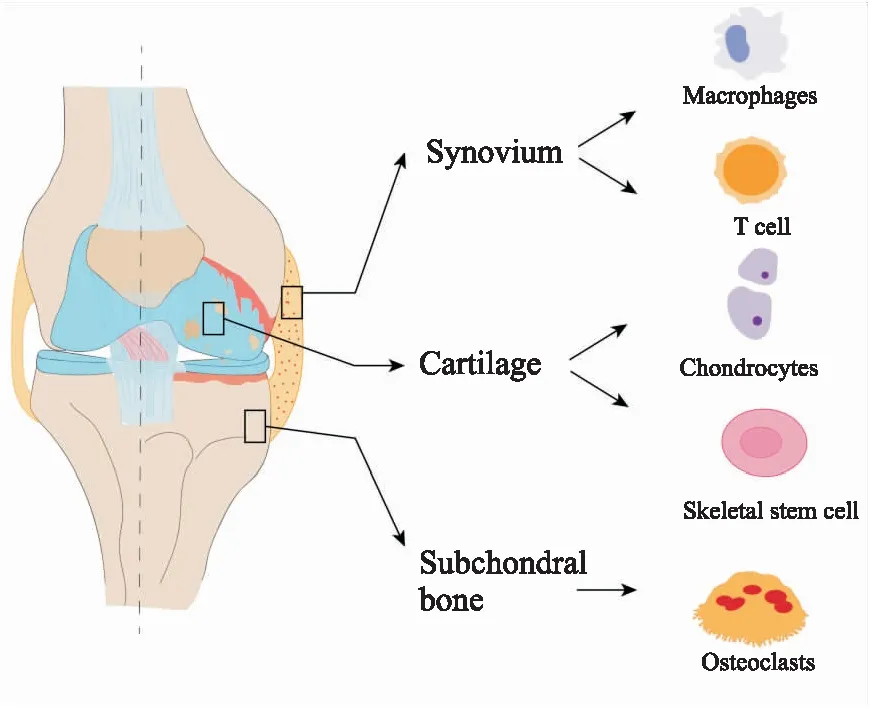
Fig 1 The main pathogenic cells and pathways involved in OA
关节软骨是一种特殊类型的透明组织,覆盖在关节的骨骼表面,有利于关节表面的滑动,并改善运动过程中的摩擦。关节软骨由软骨下骨滋养,具有抗压力的作用。软骨细胞是软骨中唯一存在的细胞类型,嵌入在主要由胶原组成的无定型细胞外基质中[3]。关节软骨由于其结构较为单一导致其自然修复能力较差。OA的明显特点之一就是关节软骨的丢失。
软骨下骨重构、骨赘形成是OA的重要特征。关节软骨和软骨下骨在一个功能单元中协同工作[4]。在OA中可以观察到破骨细胞变化导致的软骨下骨硬化和伴随着新生血管形成。软骨下骨血管生成增加,血管侵入无血管软骨,是OA的早期诊断特征[5]。
在正常生理条件下,滑膜是由巨噬细胞和成纤维细胞表型特征的薄层细胞组成,这些细胞与结缔组织形成复杂结构,在OA的发生发展中发挥重要作用,其主要功能是在关节中产生和保留滑液。在OA患者中,通常会观测到以滑膜炎为特征的炎症症状,巨噬细胞和淋巴细胞浸润,多种细胞共同作用,并产生滑膜增生、新血管生成和纤维化等症状,一般认为其与疼痛和关节功能障碍相关[6]。
骨骼中的每种组织类型的产生和维持都离不开干细胞的精准调节。其中在骨生物学领域,研究最多的是造血干细胞,但骨骼组织中还存在另一类干细胞,能够分化产生如骨、软骨等。最初人们只能在培养基中分离出骨骼间充质干细胞,直至2015年Murphy MP等[7]通过谱系追踪和克隆分析等手段,找到了骨骼干细胞(skeletal stem cell,SSCs),关节软骨中存在的SSC与OA的发生发展密切相关。
2 骨关节炎的发病机制
2.1 OA发病机制之一—软骨细胞相关的改变软骨丢失的直接原因是软骨细胞的合成代谢和分解代谢的失衡,而失衡是由多种因素引起的。而其根本原因包括衰老、创伤、肥胖、生物力学、生物节律改变等,这些原因引起的软骨细胞的肥大或凋亡、代谢障碍、细胞衰老等破坏软骨稳态,引发OA[8]。其中生物节律对软骨的代谢稳态造成影响是由于软骨细胞中的自主生物钟,这可能与软骨生物学和病理学的关键方面有关。因此,昼夜节律紊乱可能会损害组织稳态并增加对关节损伤或疾病的易感性[9]。
2.1.1软骨细胞肥大化和凋亡 软骨细胞的凋亡和肥大是软骨内骨形成过程中的自然发生的过程。健康成熟的软骨细胞在有丝分裂后保持静止状态,并通过一些机制抵抗增殖和肥大。然而,在OA患者的软骨中,可以在退行性软骨细胞中观察到肥大分化以及细胞凋亡和钙化[10]。
软骨细胞肥大以Runt相关转录因子2和人基质金属蛋白酶-13(matrix metallopeptidase 13,MMP13)的高表达为主要特征,其同时具有透明软骨标志物,如蛋白聚糖、Ⅱ型胶原蛋白和SRY-Box转录因子9等减少的特点[10]。尽管在骨骼的生长和发育过程中需要软骨细胞向肥大方向演变,但在OA病理状态下,软骨细胞的肥大是疾病进程中的不利因素。甲状旁腺激素相关蛋白能够通过抑制软骨细胞肥大来调节软骨内成骨[11]。
一些研究表明,在OA的早期发展过程中,软骨细胞的线粒体功能紊乱,会导致过量的活性氧(reactive oxygen species,ROS)产生。ROS不仅诱导氧化损伤[12],还通过激活磷脂酰肌醇-3-激酶(phosphatidylinositol-3-kinase,PI3K)/蛋白激酶B(protein kinase B,Akt)和Caspase途径诱导软骨细胞凋亡[13]。大部分的研究也证明抑制软骨凋亡可以防止OA的发生[14]。
2.1.2软骨细胞代谢障碍 在OA中,会出现低度炎症,而异常的软骨细胞代谢是对炎症微环境变化的一种反应。在OA条件下,软骨细胞经历代谢途径转变,倾向于通过从一种代谢途径转移到另一种代谢途径来适应微环境变化,例如从氧化磷酸化到糖酵解[15]。通过调节葡萄糖转运蛋白(glucose transporters,GLUT1)的表达,正常软骨细胞可以适应细胞外葡萄糖浓度的变化,而GLUT1的过表达会增加葡萄糖摄取和产生过量晚期糖基化终末产物,最终降解软骨[16]。
此外,脂质代谢调节也参与了OA发病。通过靶向脂质组学揭示了骨关节滑液中存在炎症消退标志物,包含n-3多不饱和脂肪酸的氧化产物[17]。此外,胆固醇代谢也被认为和OA调节相关,其中胆固醇-25-羟化酶-细胞色素P450家族7亚家族B成员1-视黄酸相关的孤儿受体α轴是OA发病机制的关键分解代谢调节剂[18]。我们之前的研究中发现,尿苷代谢也与OA的发生发展密切相关,关节积液中尿苷含量降低会加速软骨代谢失衡,包括软骨合成代谢减少,分解代谢增加[19]。
2.1.3软骨细胞的衰老 由衰老细胞产生衰老相关分泌表型(senescence-associated secretory phenotype,SASP)即粒细胞-巨噬细胞集落刺激因子、白细胞介素1(interleukin-1,IL-1)、白细胞介素6(interleukin-6,IL-6)、白细胞介素7(interleukin-7,IL-7)、白细胞介素8(interleukin-8,IL-8)、单核细胞趋化蛋白-1、单核细胞趋化蛋白-2、人基质金属蛋白酶1(matrix metallopeptidase1,MMP1)、MMP3、MMP10、MMP13、TIMP-2等,已在OA相关组织中发现。衰老细胞分泌的SASP可以改变组织微环境并损害干细胞或祖细胞诱导的组织再生,并导致邻近细胞的衰老,最终诱发OA,包括疼痛、活动能力受损以及形态和组织学变化等症状[20]。衰老细胞在关节软骨和滑膜中聚集,选择性清除这些细胞可以减缓创伤后OA的发展,减轻疼痛,促进软骨合成[21]。
2.1.4骨骼干细胞的耗竭 Murphy MP等[7]通过Rainbow报告基因小鼠评估了不同年龄小鼠骨关节表面克隆性骨骼生成的变化,发现随着年龄的增长,SSC数量减少,SSC活性的改变造成了OA随着年龄变化发病率升高。而通过驻留SSC的特异性激活可以促进关节软骨再生。
2.2 OA发病机制之二——软骨下骨重塑异常的软骨下骨血管生成与成骨相结合可能导致软骨下骨髓病变的发展、软骨下骨板厚度增加和最终的软骨损伤。软骨下骨中升高的血小板衍生生长因子会刺激血管生成,有助于OA的发展[22]。另外软骨下骨中升高的环氧酶2(cyclooxygenase 2,COX2)表达诱导OA相关的关节软骨退变。通过抑制COX2的表达可能抑制关节炎患者的关节破坏[23]。
近年来,越来越多的证据证明,破骨细胞启动软骨下骨重塑,深入了解破骨细胞在软骨下骨重塑中的作用将有助于发现可能的分子靶点和开发OA治疗的新策略。核因子κ B受体活化因子配体(receptor activator of nuclear factor-κ B ligand,RANKL)能够诱导单核细胞/巨噬细胞谱系细胞分化为破骨细胞,RANKL/RANK相互作用激活了一系列下游信号通路。软骨中的RANKL将破骨细胞募集到软骨下板处,释放MMP和组织蛋白酶K(cathepsin K,CTSK),从而导致局灶性软骨下骨和透明软骨的退化以及钙化软骨微裂纹的形成[24]。
2.3 OA发病机制之三——滑膜免疫细胞参与
2.3.1滑膜中的巨噬细胞 巨噬细胞是滑膜最丰富的免疫细胞之一,其在维持健康滑膜组织的稳态中发挥重要作用。活化的巨噬细胞存在于大多数(76%)的OA患者的膝盖中,其数量与OA严重程度及OA关节症状相关联,例如膝关节疼痛、关节间隙变窄和骨赘[25]。在OA发展过程中,滑膜中的巨噬细胞失去其稳定状态,以多种不同的方式被激活[26]。一种方法是激活巨噬细胞表面识别受体,进而启动细胞内包括核因子NF-κB、雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)、c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)、PI3K/Akt通路等信号通路[27],导致细胞因子和趋化因子分泌。抑制NF-κB信号通路可以将巨噬细胞的极化表型从经典活化的巨噬细胞(classically activated macrophage,M1)亚型转变为替代性活化的巨噬细胞(alternatively activated macrophage,M2)亚型,对OA起到有效的治疗作用。另一种方法是通过炎症小体介导的途径,细胞质中形成核苷酸结合寡聚化结构域样受体蛋白3(NOD-like receptor protein 3,NLRP3)炎症小体,从而产生炎症反应[26]。
其中巨噬细胞通过分泌各种介质调节关节炎症的研究比较广泛。M1巨噬细胞被干扰素-γ(interferon-γ,IFN-γ)、脂多糖、或肿瘤坏死因子α(tumor necrosis factor α,TNF-α)激活,分泌大量促炎因子,如IL-1β、IL-6、IL-12、TNF-α、IFN-γ和其他组织损害信号[26]。这些物质在典型的关节炎症和软骨基质破坏中发挥重要作用。这些因子由滑膜产生,并通过滑液扩散到软骨中,导致软骨细胞的凋亡增加,进一步加重了OA的疾病进程。同时,在近期的研究中,许多新型蛋白参与巨噬细胞调控OA滑膜炎症,例如在OA患者的滑膜组织中的巨噬细胞中发现M1极化的巨噬细胞部分大量积累,部分原因是人类新血小板反应素2的分泌[28]。随着对巨噬细胞的研究的进一步深入,关于巨噬细胞的观点一直在发生改变,巨噬细胞极化可以跨越传统公认的二元状态(即促炎M1型与抗炎M2型),并且可能涵盖广泛的表型,M1与M2的简单定义可能无法准确描述细胞的复杂性以及它们的功能。同时,科研人员也对巨噬细胞的新功能有一定程度的探索,其中Culemann等[29]通过使用命运映射方法结合三维光片荧光显微镜和单细胞RNA测序,对健康和OA关节内巨噬细胞亚群的组成、起源和分化进行全面的时空分析后发现,C-X3-C基序趋化因子受体1(C-X3-C Motif Chemokine Receptor 1+,CX3CR1+)巨噬细胞形成致密的物理屏障,将关节腔与周围的滑膜隔开,从而控制炎症的发生以保护关节内结构。全面了解不同的巨噬细胞群在OA发病和进展中的作用,使巨噬细胞调节的方法成为增强软骨修复,预防或治疗OA的新的治疗策略。
巨噬细胞也会通过与其他免疫细胞的相互作用来发挥功能。中性粒细胞群和巨噬细胞群在OA发病机制和恶化中的协同作用,会加重OA疾病进展[30]。
2.3.2滑膜中的T细胞 与健康滑膜相比,OA滑膜具有丰富的T细胞群。在OA滑膜中,T细胞的数量仅次于巨噬细胞,可能占炎症细胞的20%~25%。相较于白细胞分化抗原8阳性T细胞,白细胞分化抗原4阳性T细胞在滑膜内相对富集。多项研究发现,OA发病过程中T细胞的变化,其中Th1细胞、Th9细胞、Th17细胞、Tfh细胞和细胞毒细胞在OA患者外周血中的数量高于健康对照。辅助性T细胞在OA患者外周血中的数量低于健康对照,Th22细胞目前在OA中的确切作用尚未确定[31]。多种因素参与到T细胞对OA疾病进程的影响中,包括细胞外刺激,如外来抗原,细胞内信号,如mTOR复合物1[32]和细胞代谢,氨基酸代谢[33],操纵这些因素可能改变OA的疾病进程。
2.3.3滑膜中的其他细胞 滑膜纤维化(synovial fibrosis,SF)通常出现在OA的后期,目前的观点认为滑膜炎症驱动了纤维化的发展。大量证据表明,SF是导致关节僵硬、滑膜增生和功能受限的最重要原因之一,是中重度OA的常见症状。成纤维细胞样滑膜细胞是滑膜纤维化的主要效应细胞。由多种不同信号激活的成纤维细胞在这一过程中发挥核心作用,包括衰老、缺氧、细胞外基质变化等因素[34]。
此外,在关节中,脂肪细胞和免疫细胞之间的串扰维持关节组织稳态,导致稳态失衡也是OA的重要诱因。脂肪细胞可以产生促炎细胞因子(TNF-α、IL-6和IL-1β)和脂肪因子(瘦素和脂联素)来调节调节软骨中的炎症免疫反应[35]。
总的来说,骨关节炎发病机制主要涉及软骨、软骨下骨、滑膜等部位,软骨细胞、破骨细胞、巨噬细胞、T细胞等细胞。如Fig 2所示,在OA软骨细胞中,肥大软骨细胞增多,产生SASP的衰老软骨细胞增多以及SSC的耗竭都是导致OA的重要原因。软骨下骨中异常的血管生成和RANKL诱导破骨细胞产生的软骨下骨重塑都是OA的重要特征。在滑膜中,NF-κB、mTOR、JNK、PI3K/Akt刺激产生的巨噬细胞从M2亚型转变为M1亚型,产生IL-1β、IL-6、IL-12、TNF-α、IFN-γ和其他组织损害信号,在软骨损伤中发挥重要作用。CX3CR1+巨噬细胞形成致密的物理屏障的丢失也是OA发生的原因之一。滑膜中的T细胞、脂肪细胞等也在维持关节组织稳态中发挥重要作用。
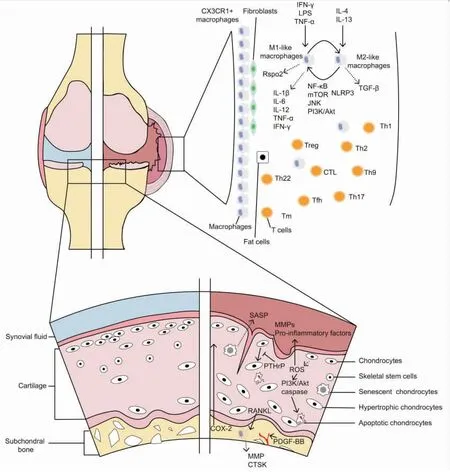
Fig 2 The molecular mechanism of OA
3 OA的靶向药物
在全球范围内,OA的患病率逐年增加,造成了巨大的社会经济负担,然而很遗憾的是目前骨关节炎的治疗手段十分有限。目前还没有改变OA进程的药物,在治疗策略中,广泛采用塞来昔布等非甾体抗炎药等仅用于消炎镇痛,并且存在肠胃道副作用和心血管疾病风险。晚期患者则只能通过关节置换术、膝盖截骨术等手术疗法。人工置换的关节虽然部分恢复关节功能,但是费用昂贵,并且存在一定的风险,如感染风险、下肢深静脉血栓形成、肺栓塞、假体周围骨折等。若能够通过药物治疗方法解决OA问题而避免手术方案,将为OA患者带来巨大福音。近年来,有关OA的新型治疗方案备受关注。OA的药物治疗应旨在减轻疾病负担(症状效应),以及通过减缓或停止潜在组织损伤的生物学过程(结构效应)来改变疾病的进程。目前国际指南推荐的治疗OA的药物治疗仅仅是对症治疗,对于根本性的治疗OA还没有更好的药物方案。对于能够改变OA疾病进程的药物研究,是全球骨科科学家和药物学家研究的重中之重。迄今为止,进入临床实验阶段的OA药物如Tab 1所示。目前关于OA的药物研究主要分为以下三种思路[36]。
3.1 靶向软骨破坏或促进OA软骨修复的药物软骨损伤是公认的OA药物的主要治疗目标。第一种策略是抑制基质降解酶表达来抑制软骨损伤。然而,遗憾的是,MMP抑制剂(例如PG116800)用于OA患者中,出现了肌肉骨骼不良的症状,这可能与MMP抑制剂可能会干扰软骨外的基质蛋白周转相关[37]。目前认为聚集蛋白聚糖酶抑制可能是比靶向胶原酶更安全的选择。自1999年被发现以来,蛋白聚糖酶,特别是ADAMTS-5,已被广泛研究作为改善疾病的OA药物开发的靶点[38]。GLPG1972/S201086是作为ADAMTS-5的小分子抑制剂,已被证明对OA小鼠和大鼠模型中的软骨具有保护作用,已经进行临床Ⅱ期试验[36]。近年来,越来越多的OA新型疗法被发现。例如,研究发现利用药物手段清除衰老的软骨细胞,以避免衰老的细胞积累,对改善OA症状具有显著效果[39]。具体来说UBX0101能够减少OA的前交叉韧带重建手术模型中的软骨损伤和关节疼痛,并刺激了人类OA软骨外植体中的软骨形成,通过促进软骨基质的合成也可以用于逆转OA进程[21]。Sprifermin通过激活成纤维细胞生长因子3(fibroblast growth factor3,FGF3),参与健康和受损软骨的软骨形成、软骨细胞增殖和软骨修复,同时刺激软骨基质产生[40]。此外,Wnt通路在OA疾病中发挥了复杂的作用。例如几种Wnt蛋白,如Wnt8a,可以激活软骨分解代谢,并且Wnt信号通路抑制剂lorecivivint,被认为具有促进软骨再生的作用,目前在进行临床Ⅲ期试验[41],而其他蛋白,如Wnt16具有软骨保护作用。
3.2 靶向OA软骨下骨重建的药物临床前的研究结果表明,在OA早期阶段,破骨细胞介导的骨吸收增强,骨密度短暂降低,但疾病后期会出现骨细胞活性减弱而出现软骨下骨硬化[42]。靶向软骨下骨破骨细胞的治疗手段可以缓解患者最关心的疼痛症状,因为它们可以通过产生轴突导向因子-1来调节感觉神经支配[43],故靶向软骨下骨的药物能够有效减轻症状。CTSK是一种主要由破骨细胞和滑膜细胞产生的蛋白酶,能够切割Ⅱ型胶原蛋白,是OA中的一个治疗靶点。口服CTSK抑制剂MIV-711可以限制膝关节OA的软骨损伤和软骨下骨重构[44]。
3.3 靶向OA滑膜炎症的药物滑膜组织中出现炎症是OA疾病的重要指标,多种因素产生的促炎因子最终导致滑膜炎的产生,并引发疼痛。糖皮质激素是世界上应用最广泛的抗炎药物,全身或关节内注射的皮质类固醇可在短期内改善OA症状,尤其是在疾病发作的情况下,但皮质类固醇注射对膝关节疼痛和功能只有短期影响,长期效果不佳[45]。
另外针对促炎因子的抗体也是一种治疗思路,尤其是抗IL-1β和抗TNF-α,已在进行了研究,但并未改善患者的症状,需要进一步研究证实[46]。除了IL-1β和TNF-α,其他细胞因子也被研究作为OA的潜在治疗靶点,例如IL-6。血清中IL-6的浓度升高与OA的发病率,并且随着时间的增加,会伴随软骨损伤[47]。其中Tocilizumab是IL-6的中和抗体,被认为可以改善手部OA的症状,已经开始进行临床III期试验。C-C基序趋化因子配体17(C-C motif chemokine ligand 17,CCL17)是单核细胞和巨噬细胞中生物效应的重要介质[48],目前正在OA患者中研究抗CCL17抗体的作用。其药物GSK3858279正在进行临床Ⅱ期试验。
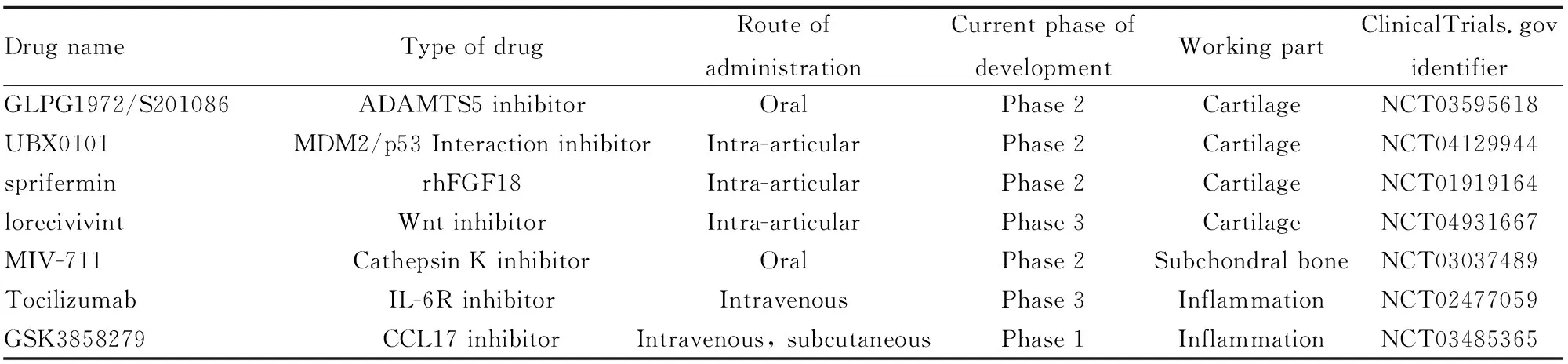
Tab 1 Main emerging therapies being investigated in OA clinical trials
随着研究的深入,关于OA病理生理学的概念仍在演变,从被视为软骨局限到影响整个关节的多因素疾病,涉及关节软骨、软骨下骨、韧带、滑膜和关节周围肌肉的结构改变。局部和全身因素之间的复杂关系调节其临床和结构表现,导致最终关节的破坏。药物治疗主要与缓解症状有关,虽然一些新的治疗干预措施的有效性和安全性被证实,但是目前还没有得到管理机构批准的治疗疾病的药物(即除了减缓或阻止疾病进展之外,还能减轻症状的治疗)。在过去的几十年里,在基础和临床研究方面的大量投入获得了与OA发展和进展相关的危险因素的重要线索。这些OA病理生理学的新发现使人们能够更好地了解疾病过程,并确定潜在的治疗靶点。未来,从机制和结构的角度了解OA疾病的最新进展将有助于开发更有效的治疗措施。
