乳腺癌新辅助化疗前后肿瘤相关巨噬细胞相关基因变化的生物信息学分析
2022-06-08陈娟蒋斌黄果贺秋冬
陈娟,蒋斌,黄果,贺秋冬
(南华大学附属第二医院1.放疗科2.整形美容科,湖南 衡阳 421001;3.南华大学衡阳医学院肿瘤研究所,湖南 衡阳 421001)
乳腺癌发病率约占所有女性癌症的30%,病死率为15%[1]。化疗已被证明可通过诱导I 型干扰素(Interferon type I,IFN-I)反应增强抗肿瘤免疫力[2-3],可以激活抗肿瘤免疫细胞并重新编程炎症性肿瘤的微环境[4]。肿瘤相关巨噬细胞(tumorassociated macrophages,TAMs) 是肿瘤微环境中炎症浸润的一种主要细胞成分,通常具有抗炎表型(M2),可促进肿瘤生长和免疫抑制。研究[5]报道乳腺癌化疗后肿瘤中产生的巨噬细胞可以通过NF-κB介导的细胞因子,包括IL-6、IL-8 和TNF-α 促进肿瘤的进展,还可通过STAT1 介导的细胞因子,包括CXCL9、CXCL10 和IL-15,促进抗肿瘤免疫。肿瘤相关免疫细胞,包括B 细胞、T 细胞、树突细胞、巨噬细胞、中性粒细胞、单核细胞和肥大细胞,可以调节癌症进展,为肿瘤的治疗提供新靶点。新辅助化疗(neoadjuvant chemotherapy,NAC)在乳腺癌的治疗中发挥着重要作用,可以减瘤降期,增加保乳几率,还可能通过消除远处隐匿性微转移改善患者总生存[6]。近期研究[7-8]表明,NAC 不仅通过对肿瘤细胞的直接细胞毒性发挥其临床作用,还通过增强恶性肿瘤微环境中的抗肿瘤免疫力来发挥作用。本文通过GEO 数据库下载乳腺癌化疗前后数据集进行分析,明确化疗前后患者TAMs 变化,评估化疗对乳腺癌患者免疫影响,为后期免疫治疗提供理论思路。
1 材料与方法
1.1 TAMs相关数据下载及分析
通过GEO 数据库[9](https://www.ncbi.nlm.nih.gov/gds) 输入Breast Cancer,TAMs,Chemotherapy 进 行检索, 通过比对, 选择人乳腺癌组织的GSE134600[5]数据集进行分析,其中包含4 对NAC 前后的乳腺癌样本数据。
1.2 差异基因筛选
通过使用R 软件(version 3.6.3)中affy 包[10]获取数据集基因表达矩阵数据,使用Normalize 函数对该数据集差异表达基因数据进行归一处理,通过R 包(limma 函数)筛选化疗前后乳腺癌组织样本的差异表达基因(differentially expressed genes,DEGs),筛选出|logFC|≥1 且校正后P<0.05 的DEG,logFC≥1 为上调基因,logFC≤-1 为下调基因。
1.3 DEGs功能富集及通路分析
使用DAVID 来分析GO 功能富集和KEGG 通路分析[11],使用“ggplot2”R 包对富集分析进行可视化。
1.4 蛋白互作网络(PPI)的构建及关键基因筛选
使用STRING 数据库(https://string-db.org)在线预测并分析筛选DEGs 之间的关联性。获得数据通过Cytoscape[12]软件(3.8.0) 进行可视化。并通过Cytohubba 插件筛选hub 核心基因,通过cBioPortal(https://www.cbioportal.org)对10 个关键基因进行突变分析。
1.5 TAMs相关的免疫细胞浸润的评估
GSE134600 数据集中4 对样本数据通过CIBERSORT[13]标准,总T 细胞计算包括CD8+T 细胞、CD4+幼稚T 细胞、CD4+记忆静止T 细胞、CD4+记忆激活T 细胞、卵泡辅助T 细胞、调节性T 细胞(Tregs)和γδT 细胞的总和。总巨噬细胞为M0、M1和M2 巨噬细胞的总和。总B 细胞为B 细胞及记忆性B 细胞的总和。同时应用CIBERSORT 包进行免疫细胞浸润的评估。使用barplot 函数、pheatmap 函数、vioplot 函数、corrplot 函数分别进行可视化绘图。P<0.05 为差异有统计学意义。
2 结 果
2.1 DEGs筛选及相关性分析
将NAC 前后两组乳腺癌测序数据进行DEGs 分析,筛选出751 个DEGs(409 个上调基因和342 个下调基因),选择两组表达差异前20 位的基因绘制火山图(图1A)、热图(图1B)以及相关性热图(图1C)。

图1 NAC前后DEGs A:火山图;B:热图;C:DEGs的相关性分析Figure 1 The DEGs before and after NAC A:Volcanic map;B:Heat map;C:Correlation analysis of DEGs
2.2 差异表达基因GO 功能富集和KEGG 通路富集分析
为了明确乳腺癌化疗前后在生物功能上差异,对上述筛选的DEGs 进行功能富集分析。通过GO富集分析DEGs 的生物过程(Biological Process,BP)、细胞组分(Cellular Component,CC)和分子功能(Molecular Function,MF)。在BP 中主要富集在IFN-I 信号通路/病毒应答与防御和病毒生命周期方面;在CC 中主要富集在细胞膜的外在成分和细胞膜的细胞质侧方面;在MF 中主要富集在细胞因子受体结合、双链RNA 结合和脂肽结合方面(图2A)。KEGG 通路富集分析中,DEGs 主要富集在甲型H1N1 流感、麻疹、丙型肝炎、冠状病毒COVID-19、NF-κB 信号通路、EBV 病毒感染、NOD样受体信号通路和阿米巴病信号通路(图2B)。

图2 DEGs功能富集分析 A:GO功能富集分析;B:KEGG通路分析Figure 2 Functional enrichment analysis of DEGs A:GO enrichment analysis;B:KEGG pathways analysis
2.3 DEGs的PPI网络及关键基因突变可视化
将DEGs 导入String 平台后,按照交互评分为0.95 处理后得到节点数据并绘制PPI 图(图3)。将获得节点数据通过CytoHubba 插件筛选与乳腺癌NAC 前后TAMs 相互作用程度最高的前10 个DEGs 作为关键基因:干扰素诱导的四肽重复蛋白1(interferoninduced protein with tetratricopeptide repeats 1,IFIT1)、干扰素刺激基因15 (IFN-stimulated gene,ISG15)、黏病毒耐药蛋白1(recombinant myxovirus resistance 1,MX1)、黏病毒耐药蛋白2 (recombinant myxovirus resistance 2,MX2)、干扰素调节因子7 (interferon regulatory factor 7,IRF7)、S-腺苷甲硫氨酸基区域蛋白 2 (radical S-adenosyl methionine domain containing 2,RSAD2)、干扰素诱导的四肽重复蛋白3 (interferon-induced protein with tetratricopeptide repeats 3,IFIT3)、干扰素诱导蛋白35(interferoninduced protein 35, IFI35)、 干扰素诱导蛋白6(interferon-induced protein 6,IFI6)、干扰素诱导跨膜蛋白1(interferon induced transmembrane protein 1,IFITM1)。通过cBioPortal 进行多组学分析10 个关键基因突变情况,IFIT1、MX1 和MX2 主要发生缺失突变,IFIT1 主要发生基因深度删除,而MX1 和MX2 主要发生基因扩增。

图3 DEGs相互作用及关键基因突变 A:PPI网络图评估DEGs相互作用关系;B:10个关键基因;C:关键基因突变Figure 3 Interactions of DEGs and hub genes mutations A: PPI network diagram of DEGs corresponding proteins; B:The 10 hub genes;C:Mutations of the hub genes
2.4 乳腺癌NAC前后免疫细胞占比和含量分析
通过CIBERSORT 包对NAC 前后乳腺癌组织中免疫细胞分布情况进行发现,占比最高的10 个免疫细胞分别是:M0 巨噬细胞、活化的树状突细胞、CD8+T 细胞、活化的自然NK 细胞、活化的肥大细胞、γδT 细胞、滤泡辅助性T 细胞、记忆性B 细胞、单核细胞、M1 巨噬细胞(图4A)。对乳腺癌免疫细胞进行无监督聚类分析发现M0 巨噬细胞、CD8+T 细胞及M2 巨噬细胞含量减少(图4B),可能是影响乳腺癌NAC 前后TAMs 相关基因表达差异的主要原因。

图4 免疫细胞占比及含量 A:22种免疫细胞的比例;B:22种免疫细胞的热图Figure 4 Proportion and content of immune cells A: The proportion of 22 kinds of immune cells; B: Heatmap of 22 immune cells
2.5 乳腺癌NAC前后免疫细胞的相关分析
乳腺癌NAC 前后免疫细胞表达相关系数较大的有:呈正相关主要有M0 巨噬细胞与记忆性B 细胞(0.64)、未活化的NK 细胞与活化的CD4+记忆性T 细胞(0.91)、初始CD4+T 细胞(0.93)、活化的树突状细胞(0.73)、活化的CD4+记忆性T 细胞与初始CD4+T 细胞活化(0.94)、树突状细胞(0.67)、CD8+T 细胞(0.61)、未活化的CD4+记忆性T 细胞与M1 巨噬细胞(0.91)、γδT 细胞(0.70)、单核细胞(0.71)、活化的NK 细胞与γδT 细胞(0.69),上述正相关免疫细胞在乳腺癌NAC 前后过程中起相互协同作用。呈负相关的可见CD8+T 细胞与单核细胞(-0.85)、未活化的CD4+记忆性T 细胞(-0.68)、M2 巨噬细胞与活化的树突细胞(-0.7)、活化的树突细胞与未活化的树突细胞(-0.78)、单核细胞与CD4+记忆性T 细胞(-0.77)、嗜酸性粒细胞与滤泡辅助性T 细胞(-0.85)、记忆性B 细胞与M1巨噬细胞(-0.67)、M0巨噬细胞与未活化的CD4+记忆性T 细胞(-0.66)、M1 巨噬细胞(-0.83),以上负相关提示在免疫细胞之间相互抑制(图5)。
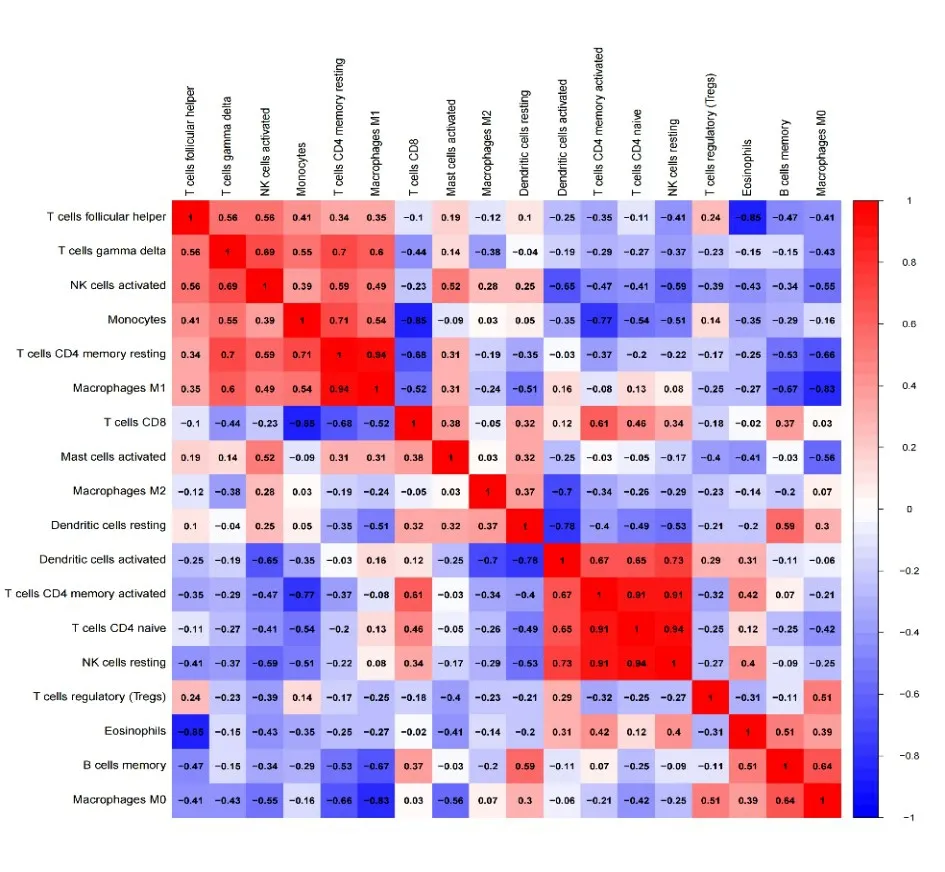
图5 免疫细胞比例的相关性Figure 5 Correlations between the proportions of the immune cells
3 讨 论
根据《2020年全球癌症统计报告》,乳腺癌已经超过肺癌,成为女性中发病率最高的恶性肿瘤,新增病例约230 万例,乳腺癌已成为癌症的第五大死因[14]。乳腺癌占女性癌症总数的30%,原发性乳腺肿瘤本身并不是乳腺癌患者的主要死因,对化疗药物的耐药性、复发性和转移性是病死率增加的主要原因。一旦转移发生,患者5年生存率只有25%[15]。尽管化疗药物具有一定的抑制特性,但一些临床证据[16]表明,化疗药物可能通过改变肿瘤免疫微环境,增强NAC 对三阴性乳腺癌的免疫作用从而发挥抗肿瘤作用,但是具体的机制及功能尚不清楚。NAC 后,残留肿瘤中肿瘤浸润淋巴细胞(TILs) 的存在可能与三阴性乳腺癌(triple negative breast cancer,TNBC) 亚型的良好预后有关[17]。在使用含阿霉素方案进行NAC 后,残留乳腺癌组织中发现T 细胞高度浸润[18]。阿霉素不仅通过招募TILs,而且通过下调PD-1 和Tim-3 检查点来增强抗肿瘤免疫[19]。
通过GSE134600 数据集筛选出NAC 前后乳腺癌组织样本中的DEGs,通过功能富集分析发现,IFN-I 信号通路在影响乳腺癌生物学过程中发挥重要作用。在胶质瘤的研究中发现,使用中等剂量间歇化疗方案可以诱导肿瘤细胞自主IFN-I 信号传导,随后释放可溶性因子,激活肿瘤细胞和肿瘤浸润免疫细胞中的干扰素刺激基因,从而激活免疫原性细胞死亡[20]。蒽环类药物在激活内体模式识别受体Toll 样受体3(TLR3)后刺激恶性细胞快速产生IFN-I,通过与肿瘤细胞上的干扰素α(IFNα) 和干扰素β 受体(IFNARs) 结合,I 型IFN-I 触发自分泌和旁分泌回路,导致趋化因子(C-X-C 基序)配体10(CXCL10)的释放,发挥抗肿瘤作用,但缺乏TLR3 或IFNα 会导致肿瘤对化疗没有反应[21]。说明IFN-I 信号通路在乳腺癌NAC 中发挥重要的免疫原性作用,诱导乳腺癌细胞凋亡。
通过CytoHubba 插件筛选出与乳腺癌NAC 前后TAMs 密切相关的10 个对应的蛋白:IFIT1、ISG15、MX1、MX2、IRF7、RSAD2、IFIT3、IFI35、IFI6、OAS2。研究[22]发现ISG15 在乳腺癌中高表达增加了乳腺癌细胞对化疗药物的敏感度。IFNα 已被用于治疗包括乳腺癌在内的各种类型癌症,但其抗肿瘤活性相对较低,阻碍了其在临床应用。目前通过将MCF-7 结合肽(MBP)与IFNα 的c 端融合,构建IFNα-MBP 融合分子(IMBP),其活性明显高于IFNα。合成的IMBP 的活性增强也与STAT1 通路靶基因(STAT1、IFIT1、IFITM1 和MX1)的激活密切相关,IMBP 可以抑制细胞周期和促进细胞凋亡途径增强了阿霉素对乳腺癌化疗的治疗效果[23]。雌激素受体阴性乳腺癌细胞对阿霉素和甲氨蝶呤的化疗诱导存在免疫休眠,主要是由于IRF7/IFN-β/IFNAR 轴的持续激活造成[24]。IRF7 上调主要通过抑制细胞主导的免疫反应转变为CD4+/CD8+T 细胞依赖性抗肿瘤反应促进乳腺癌细胞对化疗的耐药[25]。IRF7 或IFNAR 沉默可逆转休眠状态,自发摆脱休眠与干扰素-β 产生密切相关。RSAD2 在TNBC 中上调,RSAD2 表达的增加与无病生存有关[26]。
在乳腺肿瘤微环境中,TAMs 是最丰富的免疫细胞,在乳腺癌发生和进展过程中,TAMs 通过促进血管生成和癌细胞转移,诱导癌症干性,调节能量代谢和支持免疫系统抑制来支持乳腺肿瘤的生长[27]。TAMs 表现出高度的细胞可塑性,将肿瘤相关巨噬细胞重新极化为M1 巨噬细胞抑制肿瘤生长,活化的M1 和M2 巨噬细胞的不同的免疫调节功能起到肿瘤的作用[28],同时也参与肿瘤耐药的调节[29]。本研究通过免疫浸润分析,发现M0 巨噬细胞和M2 巨噬细胞在乳腺癌化疗后含量减少,而M1 巨噬细胞含量增加。相关性分析提示,M0、M2与M1 成负相关,说明化疗可能造成M0、M2 含量降低,影响患者的免疫功能。最近研究发现,成熟的树突细胞与卵巢癌患者免疫浸润和改善预后有关[30]。由SN-38 诱导的SW480 细胞上的MHC-I 类分子上调使SW480 细胞更容易受到免疫调节,从而被单核细胞衍生的树突状细胞吞噬,抑制肿瘤的转移[31]。本研究中发现活化的树突状细胞含量增加可能通过免疫调节增加乳腺癌患者的预后。研究发现在基底样乳腺癌中(basal-like breast cancer,BLBC)中IFNα、IFNγ、TNFα 通路活性明显增加,但是TGFβ 活性明显降低。IFNα、IFNγ、TNFα 通路活性与BLBC 复发呈负相关,TGFβ 通路与TNBC 复发呈正相关。BLBC 中的免疫细胞亚群分析显示M0、M1 和M2 巨噬细胞水平与TNFα 或TGFβ 通路相关,而活化记忆CD4+T 细胞的水平与这两种通路相关。此外,T 细胞亚群在具有低TGFβ 和高TNFα 通路活性的BLBC 中最为丰富。这些结果表明TNFα 和TGFβ 通路协同作用可能参与了BLBC中记忆T 细胞的调节和抗癌免疫[32]。在乳腺癌患者化疗中,顺铂可将单核细胞和M0 巨噬细胞极化为M2 样表型,加入寡聚果糖可明显减少M0 巨噬细胞极化成M2 样表型,寡聚果糖还可增加顺铂对乳腺癌细胞的毒性,防止M2 巨噬细胞分化。更重要的是,寡聚果糖抑制了肿瘤微环境中M2 巨噬细胞浸润,从而增加抗肿瘤免疫[33]。
本文通过对现有数据分析,发现乳腺癌患者化疗前有明显的免疫细胞改变,同时预测化疗前后患者体内TAMs 变化主要与干扰素信号通路密切相关,提示在乳腺癌患者化疗中可以加入干扰素进行联合处理,增加化疗药物对肿瘤细胞的杀伤效果,从而获得更优的治疗效果。但仍需对宿主、肿瘤和微环境参数行进一步研究,以阐明这些化疗与免疫之间可能存在的相关机制,以及在对乳腺癌患者的治疗中合理选择化疗、靶向药物和免疫治疗药物的组合,达到最佳治疗效果。
利益冲突:所有作者均声明不存在利益冲突。
