1个导致B1型短指的ROR2基因新突变的鉴定
2022-06-07程亚婷王陈邱雪平马建鸿马玲张铭靳冰玉赵悦张元珍郑芳武汉大学中南医院基因诊断中心检验科武汉43007湖北省产前诊断与优生优育临床研究中心武汉43007
程亚婷,王陈,邱雪平,,马建鸿,马玲,张铭,靳冰玉,赵悦,张元珍,郑芳,(.武汉大学中南医院基因诊断中心&检验科,武汉 43007;.湖北省产前诊断与优生优育临床研究中心,武汉 43007)
短指症(brachydactyly)又名短趾症,是一类由于指(趾)骨或掌(跖)骨发育异常导致的指(趾)骨缩短、缺失或者融合的家族性遗传病[1]。Temtamy和MeKusiek根据手指畸形的特征,将短指分为A~E型短指、IV型短跖、Sugarman短指、Kirner畸形8种类型[2]。B型短指症是这几种类型中最严重的一种,其根据致病基因的不同又可分为B1型(BDB1)和B2型(BDB2)。BDB1的临床表现为中间指节缩短,同时末端指节缺陷或发育不良,所有手指和脚趾均受累,拇指和大脚趾通常畸形,伴有指甲部分或完全缺失,可有不同程度的指关节粘连、软组织并指等,严重程度因人而异。BDB1呈常染色体显性遗传,致病基因是位于染色体9q22上的ROR2基因。ROR2基因全长235 kb,由9个外显子组成,编码长度为943个氨基酸的“受体酪氨酸激酶样孤儿素受体2”[3]。
我们收集了一个来自湖北武汉的BDB1家系,通过全外显子组测序及临床特征分析,确定为BDB1,致病基因为ROR2。针对候选突变位点设计引物,进行PCR扩增后Sanger测序在家系中验证该突变位点,发现了一个新的杂合突变:c.2239C>T(p.R747X),丰富了国际ROR2基因突变谱。
1 材料和方法
1.1 研究对象 先证者,女性,以“短指/趾畸形”为主诉就诊于武汉大学中南医院生殖遗传中心咨询门诊。该家系来自湖北省武汉市,汉族,4代共15人,有3人出现短指(趾)症状,符合常染色体显性遗传规律(图1)。经每位成员书面知情同意后,采集家系内3位患者Ⅲ3、Ⅰ2、Ⅱ3和2位未受累亲属Ⅱ4、Ⅱ5的外周血,并收集Ⅲ3、Ⅱ3手足部照片及X线片。本研究得到了武汉大学中南医院伦理委员会的批准(批准文号:科伦2021062)。
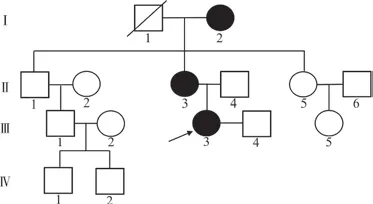
图1 BDB1家系图
1.2 外周血基因组DNA的提取 采集静脉血2 mL,EDTA-K2抗凝,采用外周血DNA提取试剂盒(Zymo Research Quick-DNATM Miniprep Plus Kit,USA)提取基因组DNA,然后进行质量鉴定,将提取的DNA产物置于-20℃保存。
1.3 全外显子测序寻找致病基因 以先证者外周血来源的基因组DNA为检测材料,首先用超声将DNA打断并制备文库,然后通过Roche KAPA HyperExome芯片对目标基因外显子及临近剪切区的DNA进行捕获和富集,最后使用MGISEQ-2000测序平台进行变异检测。测序片段通过BWA(Burrows-Wheeler Aligner)与UCSC hg19人类参考基因组进行比对,去除重复。使用基因组分析工具包(GATK,Genome Analysis Toolkit)3.7进行碱基质量值校正SNV、INDEL和基因型检测。使用ExomeDepth进行外显子水平的拷贝数变异检测。
1.4 PCR引物合成 根据全外显子测序结果,针对先证者ROR2基因第9外显子突变位点的位置,用Primer 3.0 plus设计引物,ROR2正向引物序列:5′-AGACATCTGGTCCTACGG-3′,反向引物序列:5′-ATCTGCATTGGGATCTGC-3′。引物由武汉擎科生物公司合成。
1.5 PCR扩增与Sanger测序 PCR扩增体系为50μL,采用南京诺唯赞生物科技公司PCR扩增试剂盒[Vazyme biotech,2×Taq Plus Master MixⅡ(Dye Plus)],包括25μL 2×Taq Plus Master MixⅡ(Dye Plus),正、反向引物各2μL(10μmol/L),DNA模板5μL,ddH2O 16μL。PCR反应条件:95℃预变性3 min,95℃变性30 s,60℃退火30 s,72℃延伸40 s,循环35次,72℃继续延伸5 min,4℃保温。扩增产物经20 g/L琼脂糖凝胶电泳鉴定并进行Sanger测序验证。
1.6 氨基酸保守性分析 在NCBI数据库中获取各物种ROR2蛋白的氨基酸序列文件,用DNAMA软件作图进一步可视化。
2 结果
2.1 临床特征 先证者Ⅲ3,女,31岁。双手拇指外观正常,双手第2~5指远端指节短小,第2手指尤为明显,伴有指甲发育不良(图2A);双足第2~5趾缩短,大脚趾外观正常(图2B)。X线片显示:双手第2、4~5指中远节指骨骨质融合,关节间隙消失(双手食指末节指骨稍短小),其余各掌、指骨骨质结构正常(图2C);双足第2~4趾仅见二节趾骨,余足部所示各跖、趾骨及跗骨骨质结构正常(图2D)。其母亲和外祖母表型与其类似都有短指症状,未发现其他异常(图3)。
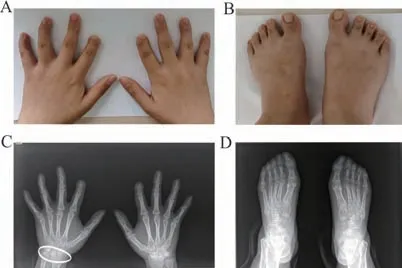
图2 先证者Ⅲ3手足部照片
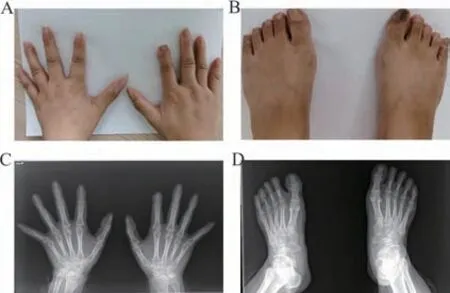
图3 先证者母亲Ⅱ3手足部照片
2.2ROR2基因突变分析与验证 通过对先证者的外周血基因组DNA进行全外显子测序,发现其ROR2基因9号外显子上存在c.2239C>T杂合突变,该突变会导致ROR2基因编码的氨基酸序列上第747位精氨酸错义终止,形成截短蛋白(图4)。根据ACMG指南标准[4],该变异被判断为致病变异(PVS1+PM1+PM2+PP1),经查询ESP数据库、人类基因突变数据库(HGMD)、EXAC外显子组整合数据库,均未见该变异报道。对家系中Ⅲ3、Ⅰ2、Ⅱ3、Ⅱ4和Ⅱ5的基因组DNA的9号外显子进行Sanger测序发现,患者Ⅲ3及Ⅰ2、Ⅱ3均存在c.2239C>T杂合突变,而未受累者Ⅱ4和Ⅱ5该位点正常(图5)。
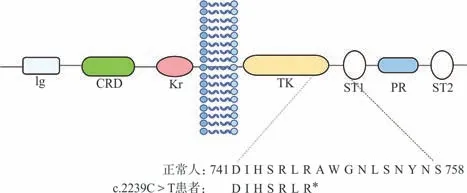
图4 ROR2蛋白模式图及预测的氨基酸变化
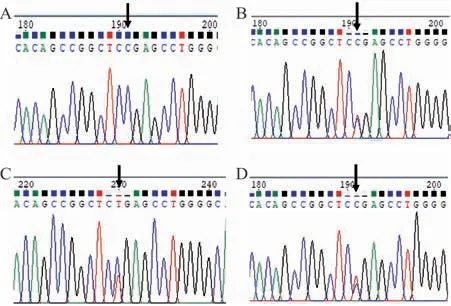
图5 ROR2基因测序图(箭头所指为突变位点)
2.3 氨基酸保守性分析 对ROR2蛋白进行氨基酸保守性分析,发现R747以及下游近30个氨基酸在多个物种中高度保守(图6)。

图6 ROR2蛋白R747及其下游保守性分析
3 讨论
B1型短指(BDB1)是一种常染色体显性遗传病,是短指中最严重的一种,其特征是手指和脚趾的末端缺失,指甲发育不良,中节指骨发育不全及不同程度的指(趾)关节粘连以及伴有拇指(趾)宽大畸形[5]。位于9q22上的酪氨酸激酶受体ROR2基因被认为是致病基因。在本研究中,通过对先证者的临床表型、X线检查、全外显子测序和一代测序验证等多方面分析,确定该先证者ROR2基因存在c.2239C>T(P.R747X)突变,依据ACMG指南标准[4],该变异被判断为致病变异(PVS1+PM1+PM2+PP1),经过对其家系进行Sanger测序验证,发现受累者均存在c.2239C>T突变,该突变在家系中与疾病存在共分离,提示ROR2基因c.2239C>T突变可能是导致该家系短指表型的原因。此突变导致编码精氨酸的密码子转变为终止密码子,从而产生一个只有747个氨基酸的截短ROR2蛋白。
为了分析R747及其上下游序列对ROR2蛋白功能的影响,我们进行了氨基酸保守性分析,发现R747以及下游近30个氨基酸在多个物种中高度保守,推测该段氨基酸序列对ROR2蛋白的正常功能非常重要。c.2239C>T突变导致R747及其下游共196个氨基酸缺失,这可能会对ROR2蛋白的功能产生重要影响。
由ROR2基因编码的受体酪氨酸激酶(RTK)样孤儿受体2属于RTK的ROR家族,由细胞外区、跨膜区和细胞内区组成。参与蛋白质相互作用的细胞外区域包含一个免疫球蛋白样(Ig-like domain,Ig)结构域,一个卷曲的半胱氨酸富集区(Frizzled-like cysteine-rich domains,CRD)和Kringle(Kringle domain,Kr)结构域。细胞内区域包括酪氨酸激酶域(Tyrosine kinase domain,TK),以及2个丝氨酸/苏氨酸富集区(Serine/threonine-rich domain,ST)和1个富含脯氨酸(Proline-rich domain,PR)的结构[6]。由于ROR2的突变产生截短蛋白的类型主要分为近端突变和远端突变。位于跨膜区之后的TK近端突变会导致整个细胞内区域的丢失[5],临床上发现这种患者的症状较轻微但表型多样。相比之下,位于TK结构域之后的远端突变产生了一个缺乏ST和PR的截短蛋白质,如我们之前所报道的c.2273C>A[7-8]等,即位于ST1的杂合突变,这种突变患者的表型往往更严重,如手指重度中远端融合,甚至手指缺失,指(趾)甲完全缺失等。少数突变是由于移码突变而产生新的C-末端,如c.2243delC(p.W749fs*24)和c.2249delG(p.G750fs*23)等,研究提示突变ROR2蛋白中额外C-末端肽的产生可能会造成并指表型[9]。本研究中发现的c.2239C>T突变发生于TK末端,产生了一个缺失TK末端以及富含丝氨酸/苏氨酸和脯氨酸结构的截短蛋白。目前国内外上发现的ROR2突变多是8、9号外显子上无义突变或移码突变导致的截短蛋白或产生的异常C末端。表1总结了国内外有关ROR2基因突变导致BDB1的突变位点。
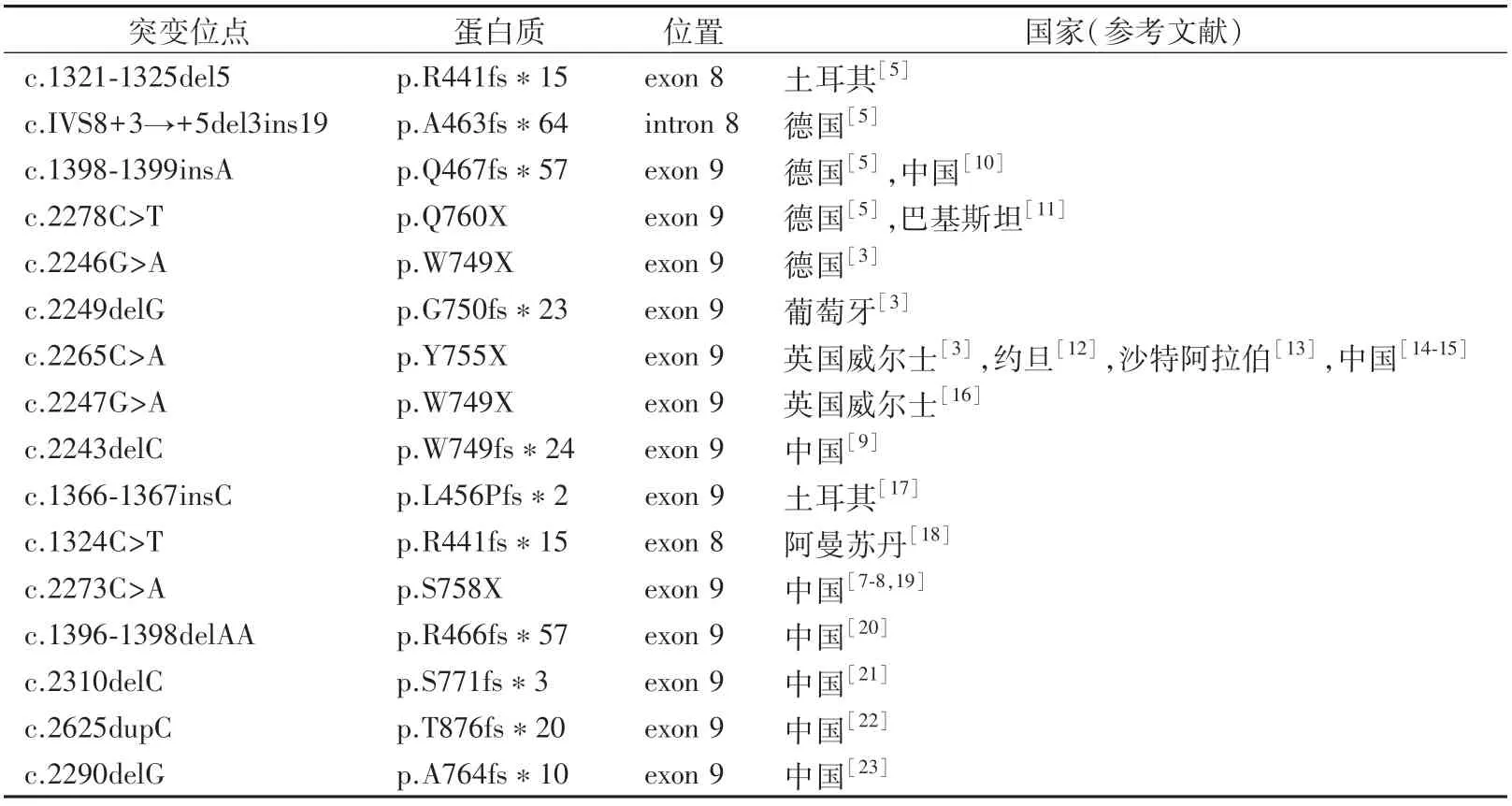
表1 导致BDB1的ROR2基因突变位点统计
由ROR2基因突变导致BDB1的分子机制并不清楚。Takeuchi等[24]发现小鼠ROR2基因杂合缺失突变没有表型。Oldridge等[3]发现两个染色体9q22杂合缺失的患者并没有表现出短指症状。这些研究说明BDB1的发病是由于ROR2特定位置突变产生了致病的蛋白,而非单纯的由于蛋白质单倍型剂量不足所致[3]。本研究报道的ROR2c.2239C>T突变导致mRNA上出现了提前终止密码子(premature termination codon,PTC),但由于该突变发生在第9号外显子上,翻译后产生了较长的多肽链,可能发生了无义介导的mRNA降解(nonsensemediated mRNA decay,NMD)逃逸,从而产生了有害的ROR2截短蛋白。研究表明,Wnt5a作为ROR2蛋白的配体,能够与ROR2的CRD结合,Wnt5a/ROR2通路可通过激活SOX9促进β-catenin降解,从而拮抗典型的Wnt信号通路参与软骨形成,在肢体骨骼发育和形态发生中发挥重要作用[25]。此外,ROR2蛋白胞内区的PR和ST2可以和细丝蛋白A(Filamin A,FLNa)相结合,从而激活Wnt5a/ROR2/JNK信号通路诱导软骨细胞的增殖和极性细胞迁移[26]。这些结果表明ROR2基因的突变可能通过上述通路信号传导异常导致BDB1的发生。
综上所述,本研究通过对1个湖北汉族BDB1家系进行突变鉴定,发现了ROR2c.2239C>T突变可导致ROR2基因编码的氨基酸序列上第747位精氨酸错义终止,形成截短蛋白。这一新的突变丰富了国际BDB1疾病的基因突变谱,结合以往我们报道的在3个湖北省BDB1家系中ROR2的另一个终止突变,提示ROR2基因的终止突变可能是湖北省BDB1疾病的主要致病基因突变。
