Extracellular vesicles from hypoxia-preconditioned mesenchymal stem cells alleviates myocardial injury by targeting thioredoxininteracting protein-mediated hypoxia-inducible factor-1α pathway
2022-06-02ChengYuMaoTianTianZhangDongJiuLiEnZhouYuQiFanQingHeChangQianWangJunFengZhang
Cheng-Yu Mao,Tian-Tian Zhang,Dong-Jiu Li,En Zhou,Yu-Qi Fan,Qing He,Chang-Qian Wang,Jun-Feng Zhang
Cheng-Yu Mao,Tian-Tian Zhang,Dong-Jiu Li,En Zhou,Yu-Qi Fan,Qing He,Chang-Qian Wang,Jun-Feng Zhang,Department of Cardiology,Shanghai Ninth People’s Hospital affiliated to Shanghai Jiao Tong University School of Medicine,Shanghai 200010,China
Abstract BACKGROUND Extracellular vesicles(EVs)derived from hypoxia-preconditioned(HP)mesenchymal stem cells(MSCs)have better cardioprotective effects against myocardial infarction(MI)in the early stage than EVs isolated from normoxic(NC)-MSCs.However,the cardioprotective mechanisms of HP-EVs are not fully understood.AIM To explore the cardioprotective mechanism of EVs derived from HP MSCs.METHODS We evaluated the cardioprotective effects of HP-EVs or NC-EVs from mouse adipose-derived MSCs(ADSCs)following hypoxia in vitro or MI in vivo,in order to improve the survival of cardiomyocytes(CMs)and restore cardiac function.The degree of CM apoptosis in each group was assessed by the terminal deoxynucleotidyl transferase dUTP nick end-labeling and Annexin V/PI assays.MicroRNA(miRNA)sequencing was used to investigate the functional RNA diversity between HP-EVs and NC-EVs from mouse ADSCs.The molecular mechanism of EVs in mediating thioredoxin-interacting protein(TXNIP)was verified by the dual-luciferase reporter assay.Co-immunoprecipitation,western blotting,and immunofluorescence were performed to determine if TXNIP is involved in hypoxia-inducible factor-1 alpha(HIF-1α)ubiquitination and degradation via the chromosomal region maintenance-1(CRM-1)-dependent nuclear transport pathway.RESULTS HP-EVs derived from MSCs reduced both infarct size(necrosis area)and apoptotic degree to a greater extent than NC-EVs from CMs subjected to hypoxia in vitro and mice with MI in vivo.Sequencing of EV-associated miRNAs showed the upregulation of 10 miRNAs predicted to bind TXNIP,an oxidative stress-associated protein.We showed miRNA224-5p,the most upregulated miRNA in HP-EVs,directly combined the 3’ untranslated region of TXNIP and demonstrated its critical protective role against hypoxia-mediated CM injury.Our results demonstrated that MI triggered TXNIP-mediated HIF-1α ubiquitination and degradation in the CRM-1-mediated nuclear transport pathway in CMs,which led to aggravated injury and hypoxia tolerance in CMs in the early stage of MI.CONCLUSION The anti-apoptotic effects of HP-EVs in alleviating MI and the hypoxic conditions of CMs until reperfusion therapy may partly result from EV miR-224-5p targeting TXNIP.
Key Words: Extracellular vesicles;Myocardial infarction;Mesenchymal stem cells;Hypoxia preconditioning;Thioredoxin-interacting protein;Hypoxia-inducible factor 1 alpha
lNTRODUCTlON
Myocardial infarction(MI)is an acute and fatal cardiovascular disease triggered by coronary occlusion,resulting ischemia-hypoxia of myocardial cells[1].Despite significant progress in surgical treatment and medical therapy,MI remains remains a major cause of morbidity and mortality in clinical practice[2].During the early period of MI,apoptosis is the predominant form of cardiomyocyte(CM)death[3].However,if reperfusion therapy cannot be initiated in time to restore blood flow,initial apoptosis transitions into passive and irreversible necrosis[4].Hence,patients with MI gain the greatest benefit from early intervention.
The molecular mechanisms underlying MI involve a double hit-related injury in CMs resulting from ischemia,hypoxia,and subsequent reoxygenation with reperfusion in infarcted tissue[5].Sustained hypoxia and excessive mitochondrial reactive oxygen species(mROS)production are common triggers of myocardial apoptosis during early MI,which occur after the apoptosome activates caspase-3[6,7].In addition,mROS and hypoxia-mediated upregulated thioredoxin-interacting protein(TXNIP)[8]interact with von Hippel-Lindau protein(pVHL)and hypoxia-inducible factor-1 alpha(HIF-1α)to promote the hypoxia-independent nuclear export and degradation of HIF-1α,hence weakening myocardial tolerance to hypoxia and eliciting anti-inflammatory responses[9].The consequent activation of TXNIP and weakened tolerance to hypoxia both contribute to the larger number of CMs undergoing apoptosis and necrosis,which are difficult to reverse during the period from MI onset to reperfusion therapy[10].Thus,due to CMs that are terminally differentiated and non-regenerative cells,the more CMs that die during the period before reperfusion,the worse the prognosis of MI[11].Accordingly,despite some mechanisms remaining obscure,interference on TXNIP have been demonstrated to protect myocardial cells from hypoxic vulnerability,programmed death,and myocardial stunning caused by ischemia[12-14].
Mesenchymal stem cells(MSCs)are a heterogeneous population of multipotent stem cells,progenitors,and differentiated cells that are existed in most stromal tissues.MSCs possess immunoregulation effects and the function of remaining internal environment stabilization and cell repair.The above-mentioned characteristics have become a cornerstone of the development of several potential therapeutic applications of MSCs,among which extracellular vesicles(EVs)derived from MSCs are the most promising[15,16].EVs are cell-derived membranous particles,which contains membranous structures of 30-2000 nm in diameter,which are packaged and secreted by most types of cells,including MSCs,and microorganisms[17].EVs mainly include exosomes and microvesicles which can regulate intracellular signal transduction by delivering proteins,mRNAs,and microRNAs(miRNAs)to targeting cells and tissues.EV carries homologous molecules from the mother cells and can adjust the biological functions of target cells,tissues,and organs,including differentiation,proliferation,migration,secretion,and death[18,19].In turn,changes in the microenvironment and physiological state of EVderived cells may influence the EV contents and their biological functions.Furthermore,a prevailing view is that the cellular origin of EVs significantly qualifies their biological function[20,21].Accordingly,EVs derived from MSCs have potential in cardioprotection[22].Thus,we determined if a preconditioned method would improve the cardioprotective effects of EVs derived from MSCs.
Because hypoxia preconditioning can enhance and strengthen the tolerance and adaptability of CMs to an anoxic environment,inflammation,and oxidative stress[23,24],the cardioprotective mechanism of EVs derived from hypoxia-preconditioned MSCs(HP-EVs)has not been fully elucidated in previous studies.We hypothesized that HP-EVs can protect CMs from ischemia and hypoxia much more effectively than EVs derived from normoxic MSCs(NC-EVs).For this purpose,EVs were extracted from mouse adipose-derived mesenchymal stem cells(ADSCs)which were pretreated with either normoxia or hypoxia preconditioning.Cardiac protection effects of HP and NC-EVs were measuredin vitroandin vivo.Additionally,we explored the molecular,morphologic and phenotypic changes with regard to MItriggered apoptosis in CMs and revealed the potential role for hypoxia-induced,EV-associated miRNAs in CM survival.
MATERlALS AND METHODS
MI and reperfusion mouse model
All animal care and procedures were approved by the Shanghai Ninth People’s Hospital Institutional Ethics Committee(Shanghai,China).Animal experimental procedures were strictly performed and followed Directive 2010/63/EU.Eight-week-old male C57BL/6 mice were purchased from Shanghai Jessie Experimental Animal Co.,Ltd.(Shanghai,China).Mice were fed standard mouse chow and waterad libitumunder specific pathogen-free conditions(20-24 °C,50%-60% humidity).All invasive procedures were performed under anesthesia.An anesthesia box with 2.5% isoflurane(RWD Life Science Co.,Ltd.,Shenzhen,China)was used to induce anesthesia for 3 min,and thereafter an animal anesthetic mask with 1.5%-2.0% isoflurane was administered in the anesthesia maintenance stage.Excess carbon dioxide inhalation was applied for euthanasia.A total of 80 healthy wild-type(WT)C57BL/6 male mice(20-24 g)were used for the experiments.The MI model was established as described by Gaoet al[25].Briefly,the left coronary artery(LCA)was ligated with a slipknot for 30,60,120,240,and 480 min to establish a time-myocardial injury relationship.Then,the slipknot was released to achieve reperfusion therapy.Successful MI was confirmed based on dynamic electrocardiograph changes(ST-segment elevation).Sham-operated mice underwent the same procedure,with the exception that the left knot on the LCA was loosened.
Transthoracic echocardiography
Echocardiography was performed to assess(M mode)ejection fraction(EF)and fractional shortening(FS)in three sequential cardiac cycles on the third day after MI surgery using echocardiography(Vevo 770 High-Resolution Imaging System;Visualsonics Inc.,Toronto,Canada).
Evaluation of area at risk and infarct size
At 12 h after loosening the knot on the LCA,the chest wall was re-opened under 1.5-2% isoflurane anesthesia to expose the heart.Then,the LCA was re-ligated and the aortic arch was clipped.Next,1%Evans Blue[normal saline(NS)as the solvent]was retrogradely injected through the ascending aorta,and the aortic arch was clipped until the non-infarction area turned blue.Then,the heart was removed and harvested,washed in NS,and sliced horizontally(parallel to the short axis of the heart)below the level of ligation.Each piece was approximately 1 mm thick.All tissue pieces were immediately incubated in 1.5% 2,3,5-triphenyltetrazolium chloride(TTC)for 20 min at 37 °C[phosphate-buffered saline(PBS)as the solvent].The infarct area and area at risk(AAR)zone were calculated by Image-Pro Plus 6.0 software.Infarct size(IS)/AAR × 100% and AAR/left ventricle(LV)area × 100% were assessed.
Isolation and culture of mouse ADSCs
ADSCs were isolated from the adipose tissue of C57BL/6 mice as previously described[26].The characterization of ADSCs was performed by flow cytometry analyses of cluster of differentiation 34(CD34),CD105,and CD106(negative controls)and CD29,CD45,and CD90(positive cell surface markers).
Isolation and culture of neonatal mouse CMs
Primary neonatal mouse CMs were extracted from approximately 150 1-day-old neonatal C57BL/6 mice as previously described[27].Briefly,75% ethanol solution was used to disinfect the neonatal mice for no more than 1 min.Then,chests were opened with the hearts quickly clipped,cut into pieces(approximate volume of 1 mm3),and placed in PBS at 4 °C.The tissues were digested in 0.125% trypsin and 0.0075 g/mL collagenase IV diluted in PBS[without fetal bovine serum(FBS)]at 4 °C overnight.The supernatant was collected and centrifuged at 200 ×gfor 5 min.Then,the tissues and cells were resuspended and cultured in complete medium[Dulbecco’s Modified Eagle Medium(DMEM)-high glucose containing 5% FBS at 37 °C,5% CO2]for 2 h to induce fibroblast attachment before CMs.The remaining supernatant(only containing CMs)was plated in new 2-cm dishes at a density of 1 × 106cells/mL for a subsequent study.α-actinin staining was applied to identify the purity of the CMs.See Figure 1A for a flow chart of the isolation procedures for ADSCs and CMs.
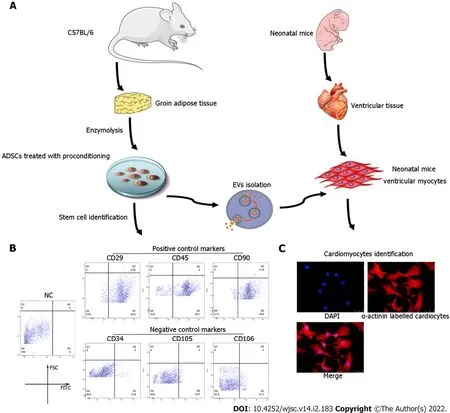
Figure 1 ldentification of adipose-derived mesenchymal stem cells and neonatal mouse cardiomyocytes.A:Flow chart of extracellular vesicles and neonatal mouse cardiomyocyte(CM)isolation;B:Characterization of adipose-derived mesenchymal stem cells(ADSCs)was performed by flow cytometry analyses of cluster of differentiation 34(CD34),CD105,and CD106(negative controls)and CD29,CD45,and CD90(positive cell surface markers);C:Identification of neonatal mouse CMs,which were specifically stained with α-actinin.CM:Cardiomyocyte;ADSC:Adipose-derived mesenchymal stem cells;CD:Cluster of differentiation.
In vitro CM hypoxia model
Anin vitromodel of mouse CM hypoxia was established by incubating cells in oxygen-free,low-glucose DMEM in a controlled atmosphere(5% CO2,95% N2)for 2 h.Then,the incubation conditions were converted to normoxia FBS-free medium for 12 h.After treatment,the myocardial cells were collected and analyzed.
Hypoxic preconditioning of ADSCs
ADSCs were seeded in complete medium(DMEM/F12 with 10% EV-free FBS)for 24 h.Oxygen-free DMEM/F12 medium previously incubated overnight with 100% N2was prepared in advance.Hypoxic preconditioning was performed by exposing the cells to five cycles of hypoxia(60 min in oxygen-free DMEM/F12 medium and 5% CO2,95% N2cultured atmosphere)with intermittent reoxygenation(30 min in normal oxygen-containing DMEM/F12 medium and 5% CO2,75% N2,20% O2cultured atmosphere)in a hypoxic chamber(Forma-1025 Anaerobic System;Thermo Fisher Scientific,Waltham,MA,United States).After hypoxic preconditioning,ADSCs were cultured in serum-free DMEM/F12 medium in a normoxic environment(5% CO2),and the supernatant was collected for EV extraction after 24 h.
Isolation and characterization of EVs
EVs were extracted from cultured ADSCs(approximately 107perdish)in the absence or presence of exposure to hypoxic preconditioning by differential velocity centrifugation.Briefly,the cell culture supernatant was centrifuged at 2,000 ×gfor 30 min at 4 °C to remove cell debris.Then,the supernatant was collected and centrifuged at 100000 ×gfor 70 min to precipitate the EVs.The supernatant was discarded to remove contaminating proteins and EVs were re-suspended in PBS.Size distribution and concentration of the EVs were determined using the NanoSight NS300 Instrument(Malvern Instruments,Malvern,United Kingdom),and EV morphology was assessed by transmission electron microscopy(TEM).Expression of the EV surface markers[tumor susceptibility gene 101(TSG101),CD63,CD81]was detected by western blotting.
EV injections
Eighty mice were randomly divided into the following four groups(n= 20 each):sham(no MI,control),MI,NC-EV(NC-EVs plus MI),and HP-EV(HP-EVs plus MI),which ensured a sample size of more than five miceperassay.In the NC-EV and HP-EV groups,EVs were administered at a dose of 1 μg/1 g body weightviainjecting into the border zone of the infarcted heart at three sites immediately post-MI surgery.
Confirmation of EV uptake by CMs
EVs were labeled with PKH26(Cat.MINI26;Sigma-Aldrich,St.Louis,MO,United States),and mouse CMs were labeled with phalloidin(Cat.A12379s;Thermo Fisher Scientific).Then,500 μL EV solution was stained with 5 mL PKH26 and added to a culture of CMs,EVs,and cells followed by a 2-h incubation to allow endocytosis by mouse CMs.The CMs were washed three times with PBS and then fixed in 4% paraformaldehyde for 20 min,after which the nucleus was stained with Hoechst.An inverted microscope was used to detect the EVs phagocytized in the CMs.
Cell transfection
HEK 293T cells were transfected with miR-224-5p mimics and the TXNIP dual-luciferase plasmid(Ribobio,Shanghai,China).H9c2 CMs stably overexpressing TXNIP and TXNIP-L294A mutant were established by lentivirus(synthesis by ZoRin,Shanghai,China).ADSCs overexpressing miR-224 and miR224-negative control(NC)were infected with adeno-associated virus(AAV)containing the miR-224 or miR-224-NC sequence(RiboBio),and miR-224 was knocked out by CRISPR/Cas9 in ADSCs(RiboBio).
Dual luciferase reporter assay
First,miR-224-5p mimics and PGL3 Luciferase plasmids containing WT,NC,or mutated TXNIP 3’-untranslated region(3’-UTR)sequences were co-transfected in HEK 293T cells,which were cultured in 24-well plates and co-transfected at approximately 70% confluence.After 12 h,the cells were re-cultured in 96-well luciferase assay plates.The ratio of firefly to Renilla luciferase activity was detected after 36 h using the Dual-GLOTMLuciferase Assay System(Cat.E2920;Promega,Madison,WI,United States).
Quantitative polymerase chain reaction and western blotting
We used the RNAiso Plus extraction reagent(Cat.9108;Takara,Dalian,China)to extract EV-associated RNA.Stem-loop primers(Ribobio Biotech)were used to generate the cDNA of miRNA.The cDNA was amplified by SYBR green-based quantitative polymerase chain reaction(qPCR).U6 small nuclear RNA was used as the internal control.Cardiac tissues and CM protein were extracted using radioimmunoprecipitation assay buffer.TSG101,CD63,CD81,TXNIP,HIF1,and ubiquitin antibodies were supplied by Abcam(Cambridge,MA,United States);α-tubulin was supplied by Cell Signaling Technology(Danvers,MA,United States).
Statistical analyses
SPSS 19.0 software was used for the data analyses(IBM Corp.,Armonk,NY,United States).Whether the data fit the normal distribution was assessed by the Shapiro-Wilk test.Categorical variables were analyzed by the Pearson’s chi-square test(n≥ 5)or Fisher’s exact test(n< 5)with subsequent multiple comparisons using Bonferroni correction.One-way analysis of variance with subsequent post-hoc multiple comparisons test(Student-Newman-Keuls test)was applied for continuous variables.The Kruskal-Wallis test was applied for nonparametric testing of multiple independent samples,and a Dunn-Bonferroni test used for post-hoc comparisons.
RESULTS
Characterization of ADSCs,ADSC-derived EVs,and mouse CMs
ADSCs isolated from mouse adipose tissue were identified by using cell surface markers of stem cells.The positive cell surface markers were CD29,CD45,and CD90 which demonstrated positive expression(> 95%)in flow cytometry assessment.Likewise,negative cell surface markers(CD34,CD105,and CD106)revealed low/negative expression(Figure 1B).Subsequently,neonatal mouse CMs were identifiedviaα-actinin staining(Figure 1C).Sequential supercentrifugation was adapted to gain EVs from supernatant of ADSCs.TEM and the NanoSight Instrument were applied to verify the isolated EVs(Figure 2A and B).Results showed that the isolated EVs had a average diameter of 115 nm.Western blot assay revealed that EVs expressed three EV-associated markers:CD63,CD81,and TSG101(Figure 2C).CMs’ endocytosis of EVs was verified by PKH26-stained EVs detectedviafluorescence microscopy(Figure 2D).
HP-EVs reduce myocardial injury in the early stage of MI and hypoxia
To access the temporal relationship of HP-EV cardioprotective effectsin vitroandin vivo,MI models were established and LCAs were ligated for 0.5,1,2,4,and 8 h followed by reperfusion for 12 h.Evans Blue/TTC staining was used to evaluate the area of viable myocardium in the HP-EV and MI groupsin vivo(Figure 3A).The results demonstrated that,within the first 2 h,HP-EVs contributed to significant cardiomyocyte survival compared to the MI group,which peaked after 1 h of LCA ligation followed by 12 h of reperfusion(HP-EVsvsMI;P= 0.0021).Similarly,the Cell Counting Kit-8 assay was used to study the temporal relationship of HP-EV cardioprotective effectsin vitro(Figure 3B).Neonatal mouse CMs were exposed to hypoxic conditions for 0.5,1,2,4,and 8 h followed by 12 h of reoxygenation.CMs treated with HP-EVs showed significant cardioprotective effects against hypoxia compared to the control group,which peaked at 2 h(HP-EVvshypoxia-CMs;P= 0.0009)and decreased with a prolonged period of hypoxia.Thus,2-h hypoxiain vitroand 1-h MIin vivowere applied to subsequent experiments.
HP-EVs fail to alleviate myocardial injury in CMs subjected to a long period of hypoxia
We evaluated the cardioprotective effects of HP-EVs in improving heart function and alleviating the degree of CM apoptosis after a long period of hypoxia and ischemia(8 h of hypoxia for CMs and 8 h of LCA ligation in mice).The cardioprotective effects of HP-EVs did not reduce apoptosis nor improve heart function after a long period of hypoxia or ischemia(P= 0.400 andP= 0.7136 for CM apoptosisin vitro,Figure 4A and B;P= 0.1519 for myocardial apoptosisin vivo,Figure 4C;P= 0.486 for EF% andP=0.785 for FS%,Figure 4D).These results indicated that the cardioprotective effects of HP-EVs might decrease with prolonged ischemia or hypoxia.Thus,in the case of late-stage apoptosis and irreversible necrosis induced by protracted and prolonged ischemia or hypoxia,ADSC-derived EVs(ADSC-EVs)contribute little to ameliorating myocardial injury,which is consistent with our conventional understanding that patients with MI gain the greatest benefit from early intervention.
ADSC-EVs reduce apoptosis in CMs subjected to hypoxia
To determine the anti-apoptotic effects of ADSC-EVs in preventing or attenuating MI-triggered apoptosis in CMs,apoptosis assays were performed by the Annexin V/PI,terminal deoxynucleotidyl transferase dUTP nick end-labeling(TUNEL),and caspase-3 activation assays in cultured neonatal mouse CMs subjected to hypoxia and were exposed to NC-EVs or HP-EVs prior to hypoxia.As is revealed in Figure 5A,pretreatment with NC-EVs decreased the apoptotic rate of CMs[P= 0.0021 compared to the hypoxia/reoxygenation(H/R)group].However,the downregulation of apoptosis was significantly higher after exposure to HP-EVs(P= 0.0080 compared to NC-EVs).Over again,the cardioprotective effects were significantly greater in HP-EV-treated cells as determined by the TUNEL assay(P= 0.0001 compared to the H/R group;P= 0.0291 compared to NC-EVs)(Figure 5B).Meanwhile,as shown in Figure 5C,caspase-3 activation assays demonstrated a reduced trend in the enzymatic activity of caspase-3 after CM treatment with ADSC-EVs,with HP-EVs eliciting more significant antiapoptotic effects(P= 0.0001 compared to the H/R group;P= 0.0001 compared to NC-EVs).
ADSC-EVs ameliorate myocardial damage in a mouse model of MI
To assess cardioprotective effects in alleviating MI-triggered myocardial injury of ADSC-EVsin vivo,in situapoptosis was evaluated in infarct tissues by the TUNEL assay.Figure 6A demonstrates that the degree ofin situapoptosis was significantly improved in the NC-EV and HP-EV groups.Moreover,the HP-EV group had a greater ameliorative apoptotic rate than the NC-EV group.Cardiac IS and AAR were evaluated in ischemic myocardium injected with ADSC-EVs post-MI models establishment.As is revealed in Figure 6B,among the sham,MI,NC-EV,and HP-EVs groups,AAR/LV values were similar(LCA was re-ligated prior to Evans Blue staining to calculate the AAR/LV values).Significantly,the NC-EV and HP-EV groups showed markedly mitigated post-MI IS compared to the ischemiareperfusion(IR)group.Once again,HP-EV group demonstrated a conspicuously mitigated IS region(NC-EVvsIR,P= 0.0115;HP-EVvsNC-EV,P= 0.0213).
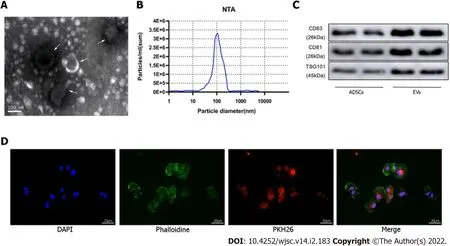
Figure 2 ldentification of extracellular vesicles.A:Transmission electron microscopy characterization of extracellular vesicles(EVs);B:Size distribution by intensity was detected using the NanoSight Instrument;C:EV biomarkers cluster of differentiation 63(CD63),CD81,and tumor susceptibility gene 101 were identified by western blotting of adipose-derived mesenchymal stem cell(ADSC)-derived EVs and ADSCs;D:EV tracer assay was used to identify exosomes phagocytosed by cardiomyocytes.ADSC:Adipose-derived mesenchymal stem cell;EV:Extracellular vesicles.
ADSC-EVs improve cardiac function after MI reperfusion injury
Echocardiography on Day 3 post-MI revealed that EF and LV fractional shortening were markedly turned better of cardiac systolic function that treated with ADSC-EVs(Figure 6C),particularly HP-EVs(EF Day 3:NC-EVsvsIR,P= 0.022;HP-EVsvsNC-EVs,P= 0.030;FS Day 3:NC-EVsvsIR,P= 0.031;HP-EVsvsNC-EVs,P= 0.017).
Hypoxia preconditioning upregulates miR-224-5p,a potential TXNIP regulator,in ADSC-EVs
To discover the molecular mechanism of much more significant cardioprotective effects of HP-EVs against ischemia and hypoxia-induced CMs damage,sequencing analysis was applied to reveal nucleic acid molecular(miRNA)expression differences between NC-EVs and HP-EVs.The result of sequencing detected 88 miRNAs expression differences between HP-EVs and NC-EVs.Further analysis revealed 10 of them were potentially predicted to bind with TXNIP by the TargetScan and miRanda algorithms in the Encyclopedia of RNA Interactomes database(Figure 7A).The expression differences of these miRNAs were verified by quantitative polymerase chain reaction(Figure 7A).Then we chose miR-224-5p as candidate for further study for its most markedly upregulated expression among the 10 miRNAs in HP-EVs.In the follow-up verification work ,we used the dual-luciferase reporter assay to determine that miR-224-5p directly bind to TXNIP WT-3’-UTR region of its mRNA to inhibit translation process of TXNIP(P= 0.0026;Figure 7B).
ADSC-EV miR-224-5p ameliorates HIF-1α degradation and hypoxia-induced CM apoptosis by targeting TXNIP
To assess whether ADSC-EV miR-224-5p can attenuate the degradation of HIF-1α and hypoxia-induced apoptosis in CMs by inhibiting TXNIP,we established AAV-miR-224 ADSCs and ADSCs with miR-224 knocked out to obtain miR-224-5p overexpressing EVs and miR-224-5p knockout EVs,respectively.TXNIP,HIF1-αexpression level and apoptotic degree of CMs were evaluated on hypoxia-treated neonatal mouse CMs pre-processed with EVs derived from ADSCs,ADSCs overexpressing miR-224,and ADSCs with miR-224 knocked out by CRISPR/Cas9.As shown in Figure 7C,pre-treatment with EVs derived from ADSCs overexpressing miR-224 Led to the significant suppression of TXNIP expression relative to NC-ADSCs(P= 0.0006)and increase in HIF-1α expression(P< 0.0001).In EVs derived from ADSCs with miR-224 knocked-out,TXNIP expression was not inhibited(P= 0.0018)and HIF-1α expression was decreased(P= 0.0002)compared to the NC group.Furthermore,Annexin V/PI staining indicated,as expected,a significant improvement in apoptosis in CMs treated with miR224-5poverexpressing EVs compared to NC-EV-treated cells(P= 0.0002;Figure 7D).This effect was significantly reversed by miR-224-5p knockout EVs compared to the NC group(P= 0.0059).
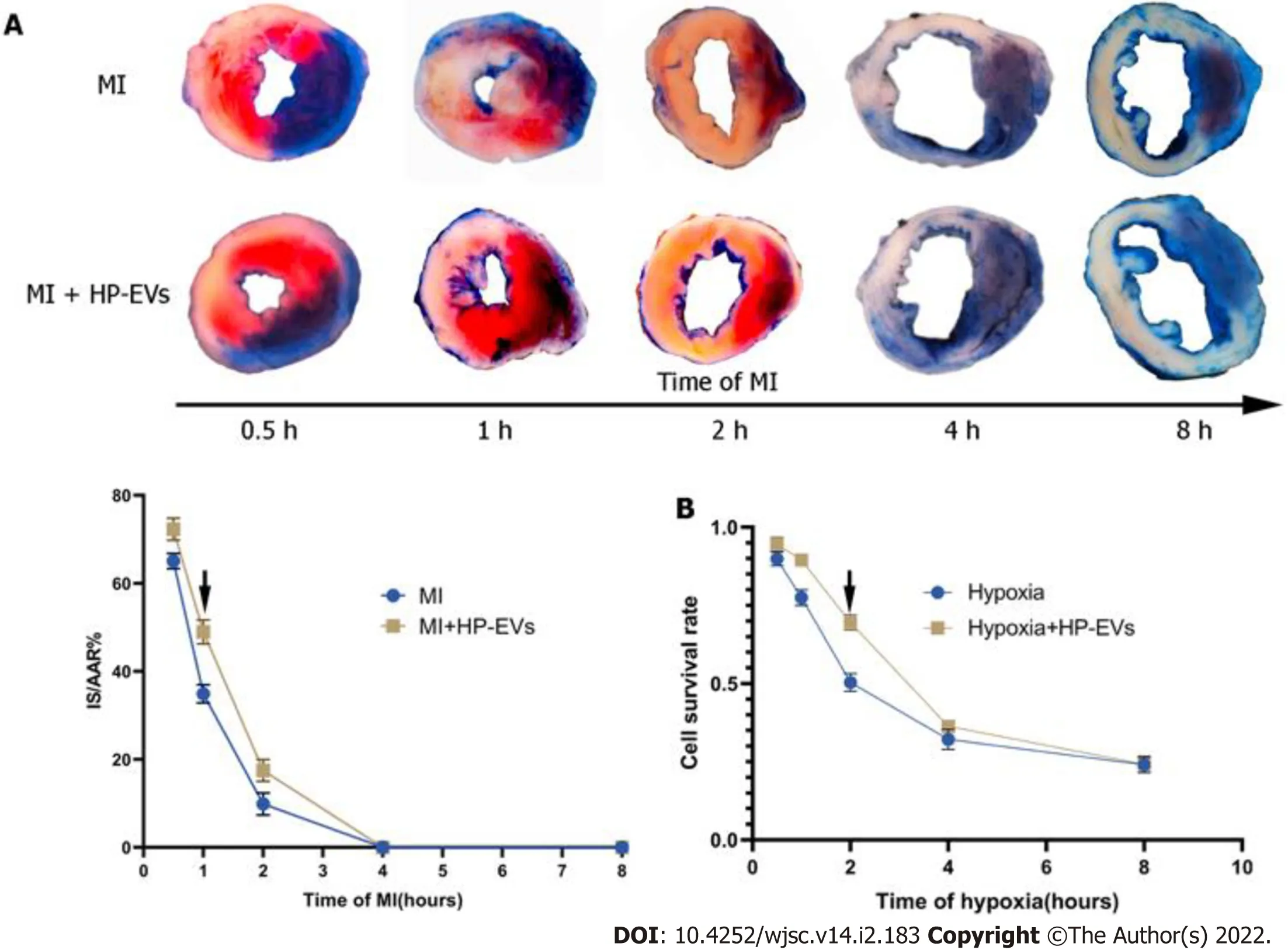
Figure 3 Evaluation of the temporal relationship of the cardioprotective effects of hypoxia-preconditioned extracellular vesicles.A:Temporal relationship of the cardioprotective effects of hypoxia-preconditioned extracellular vesicles(HP-EVs)in vivo.Myocardial infarction(MI)models were established with left coronary artery ligation for 0.5,1,2,4,and 8 h followed by 12 h of reperfusion.Evans Blue/2,3,5-triphenyltetrazolium chloride staining was used to evaluate the area of viable myocardium in the HP-EV and MI groups(n = 5);B:Temporal relationship of the HP-EV cardioprotective effects in vitro.Hypoxia injury models were established for 0.5,1,2,4,and 8 h of hypoxia followed by 12 h of reoxygenation.Cell Counting Kit-8 assay was used to evaluate the cell survival rate in the HP-EV and hypoxia groups(x axis was time of hypoxia,y axis was the value of absorbance of experimental/control group,which reflected the survival rate of cells in the experimental condition;n = 5).The arrows indicate the most significant difference,and the corresponding time points were adopted for subsequent research.MI:Myocardial infarction;HP-EV:Hypoxia-preconditioned extracellular vesicles.
TXNIP regulates the ubiquitination of HIF-1α in a chromosomal region maintenance-1-dependent manner
TXNIP binds to the β-domain of pVHL and promotes the degradation of HIF1α independently of hypoxia.A functional nuclear export signal(NES)in the chromosomal region maintenance-1(CRM-1)-binding site(Leu294)of TXNIP is important for the formation of the TXNIP-pVHL-HIF-1α complex[9].To further explore the mechanism by which TXNIP regulates HIF-1α degradation in CMs,TXNIPoverexpressing and TXNIP L294A mutant H9c2 cell lines were established(Cyagen Biosciences,Guangzhou,China).Our results demonstrated that the nuclear expression of HIF-1α was abolished in TXNIP-overexpressing cells in hypoxia.In TXNIP L294A mutant cells,the nuclear expression of HIF-1α was recovered.After treatment with leptomycin B,which specifically blocks CRM1-dependent nuclear export and is extensively used to investigate this process,TXNIP-induced HIF-1α degradation was inhibited(Figure 8A).Western blot analysis revealed that TXNIP induced the degradation of HIF-1α in H9c2 CMs in the presence and absence of hypoxia(Figure 8B).Moreover,the increased ubiquitination of HIF-1α induced by overexpressed TXNIP was detected(Figure 8C and D).However,this effect was not detected in TXNIP L294A mutant cells,which indicated that,as a functional NES binding site of CRM-1 in TXNIP,TXNIP-mediated ubiquitination and degradation of HIF-1α by the proteasome might be dependent on CRM-1-mediated nuclear transport and stabilization.
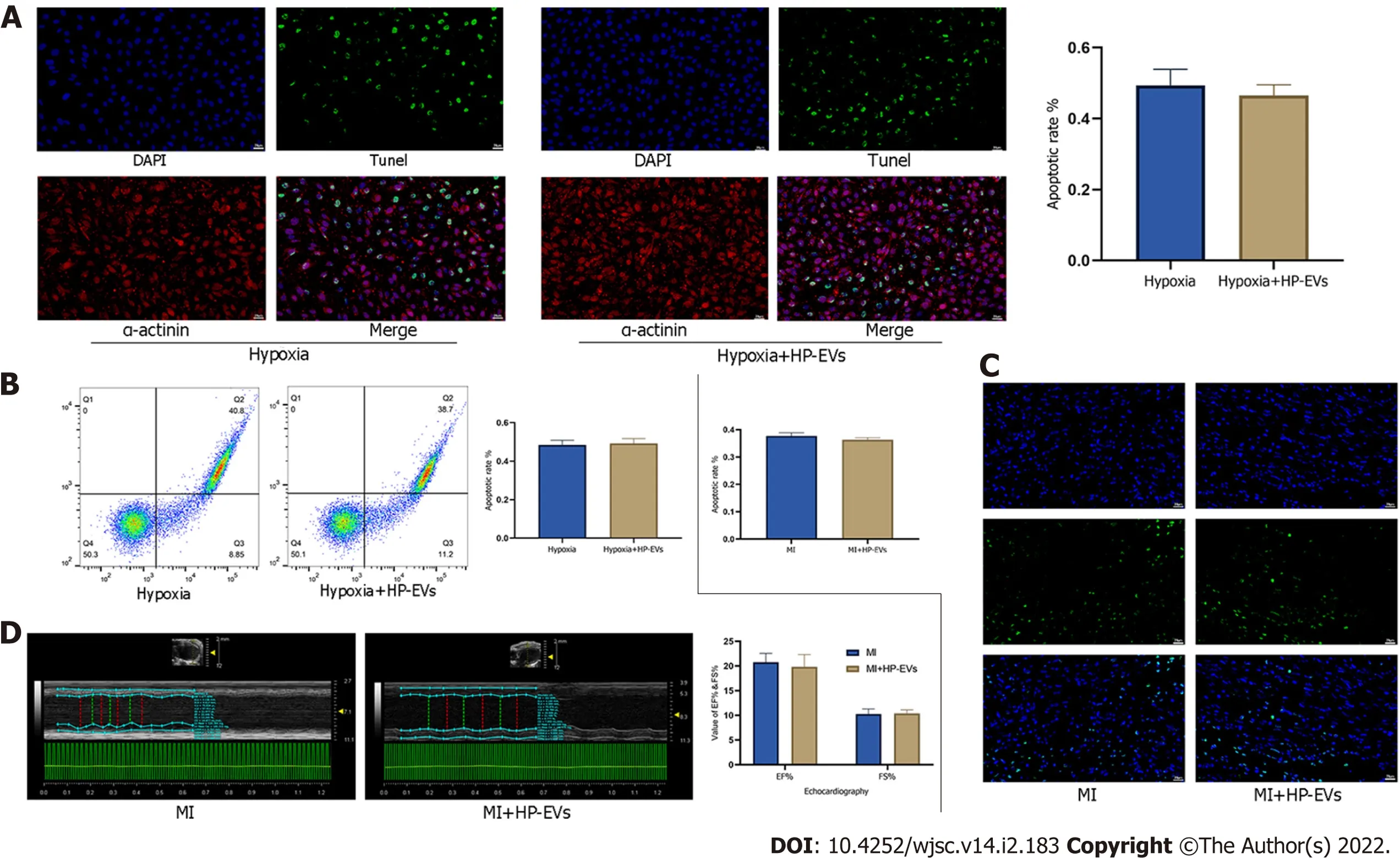
Figure 4 Cardioprotective effects of hypoxia-preconditioned extracellular vesicles after long periods of hypoxia and ischemia.A:Degree of in situ apoptosis of cardiomyocytes(CMs)in hypoxia and hypoxia-preconditioned extracellular vesicle(HP-EV)groups after a long period(8 h)of hypoxia followed by 12 h of reoxygenation,as determined by the terminal deoxynucleotidyl transferase dUTP nick end-labeling(TUNEL)assay(n = 5);B:Degree of CM apoptosis in the hypoxia and HP-EVs groups after a long period(8 h)of hypoxia followed by 12 h of reoxygenation,as determined by flow cytometry(n = 5);C:Degree of in situ apoptosis of CMs in the myocardial infarction(MI)and HP-EVs groups after a long period of MI[left coronary arteries(LCAs)were ligated for 8 h followed by 12 h of reperfusion],as determined by the TUNEL assay(n = 5);D:Echocardiography was used to examine the heart function of the MI and HP-EVs groups on Day 3 after a long period of MI(LCAs were ligated for 8 h followed by 12 h of reperfusion);ejection fraction and fractional shortening were detected(n = 5).MI:Myocardial infarction;HP-EV:Hypoxia-preconditioned extracellular vesicles.
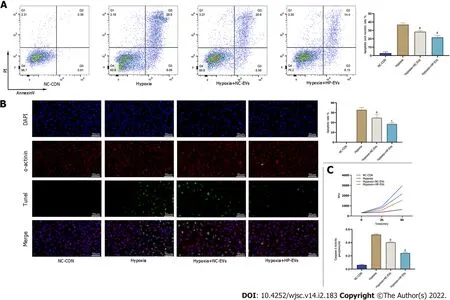
Figure 5 Hypoxia-preconditioned extracellular vesicles alleviated hypoxia/reoxygenation-induced apoptosis in vitro.A:Degree of cardiomyocyte(CM)apoptosis in control,hypoxia,normoxic extracellular vesicle(NC-EV),and hypoxia-preconditioned EV(HP-EV)groups after 2 h of hypoxia followed by 12 h of reoxygenation,as determined by the Annexin V/PI assay(n = 5);B:Degree of CM apoptosis in control,hypoxia,NC-EV,and HP-EV groups after 2 h of hypoxia followed by 12 h of reoxygenation,as determined by the terminal deoxynucleotidyl transferase dUTP nick end-labeling assay(n = 5);C:Caspase-3 activity of CMs in the control,hypoxia,NC-EV,and HP-EV groups after 2 h of hypoxia followed by 12 h of reoxygenation(n = 5).All data are expressed as the mean± SD.aP < 0.05,bP < 0.01 compared with Hypoxia group;cP < 0.05,dP < 0.01 compared with Hypoxia + NC-EVs group.HP-EV:Hypoxia-preconditioned extracellular vesicles;NC-EV:Normoxic extracellular vesicle.
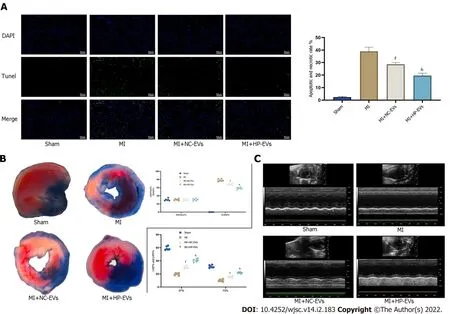
Figure 6 Hypoxia-preconditioned extracellular vesicles alleviated hypoxia/reoxygenation-induced apoptosis in vivo.A:Degree of cardiomyocyte(CM)apoptosis in the control,sham,myocardial infarction(MI),normoxic extracellular vesicle(NC-EV),and hypoxia-preconditioned EV(HP-EV)groups after 2 h of left coronary artery(LCA)ligation followed by 12 h of reperfusion,as determined by the terminal deoxynucleotidyl transferase dUTP nick endlabeling assay(n = 5);B:HP-EV cardioprotective effects were assessed in vivo,and MI models were established with LCA ligation for 1 h followed by 12 h of reperfusion.Evans Blue/2,3,5-triphenyltetrazolium chloride staining was used to evaluate the area of viable myocardium in the sham,MI,NC-EV,and HP-EV groups(n = 5);C:Heart function of the sham,MI,NC-EV,and HP-EV groups was evaluated by echocardiography;ejection fraction and fractional shortening were detected(n= 6).All data are expressed as the mean ± SD.eP < 0.05,fP < 0.01 compared with MI group;gP < 0.05,hP < 0.01 compared with MI + NC-EVs group.MI:Myocardial infarction;HP-EV:Hypoxia-preconditioned extracellular vesicles;NC-EV:Normoxic extracellular vesicle.
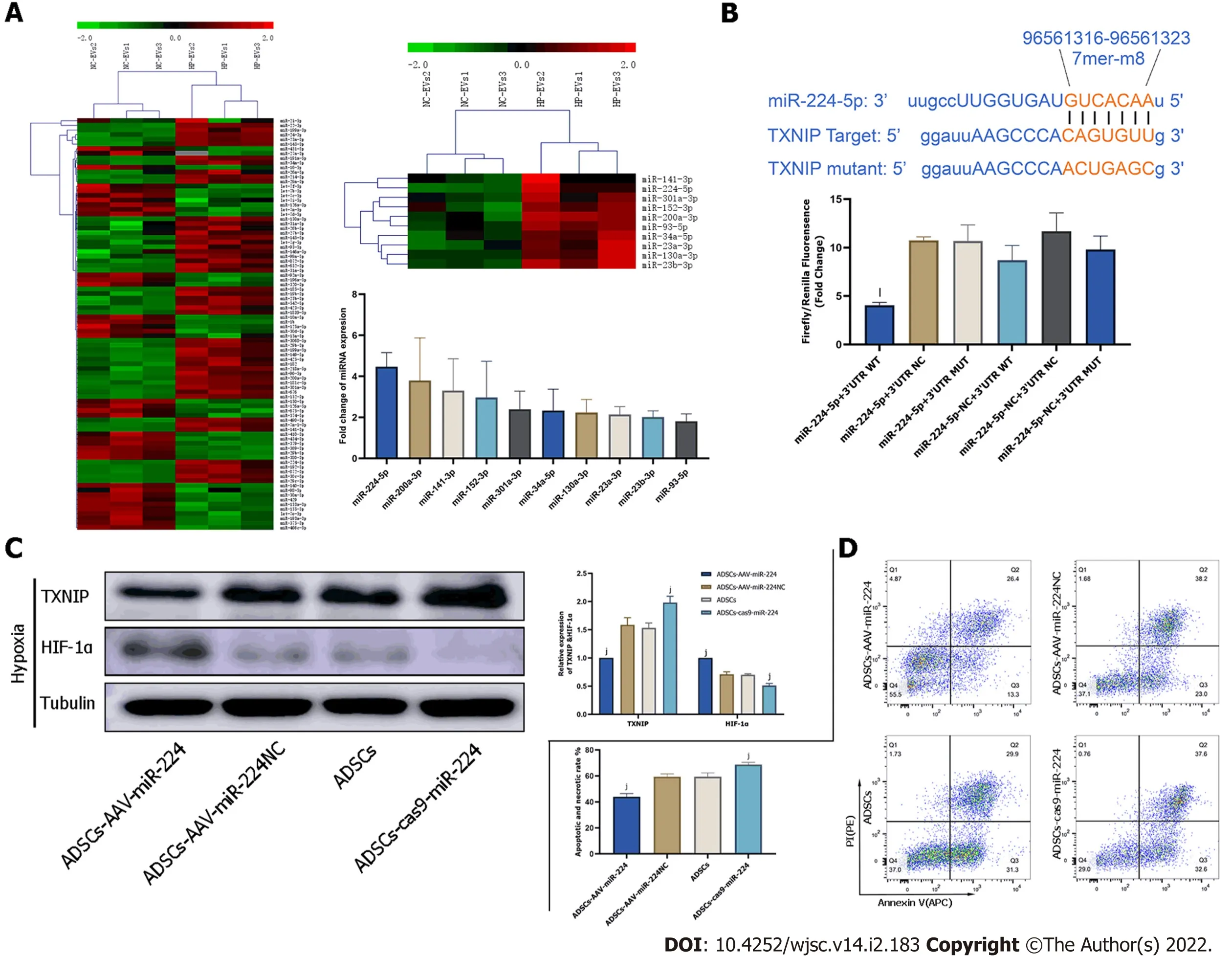
Figure 7 Extracellular vesicle-associated miR-224-5p inhibits hypoxia-induced apoptosis in cardiomyocytes by downregulating thioredoxin-interacting protein.A:MicroRNA(miRNA)sequencing analysis in adipose-derived stem cell-extracellular vesicles(ADSC-EVs).A total of 88 miRNAs were upregulated in hypoxia preconditioned EVs(HP-EVs)compared to normoxic EVs(NC-EVs),of which 10(shown in the heatmap)were predicted to associate with thioredoxin-interacting protein(TXNIP).Validation of differential EV-associated miRNA expression through quantitative PCR.U6 small nuclear RNA served as the internal reference;B:Dual luciferase reporter assay.HEK 293 T cells were co-transfected with miR-224-5p mimics and PGL3 Luciferase reporter plasmids containing wild-type(WT)or mutated TXNIP 3′-untranslated region(3’-UTR).mutated-TXNIP 3′-UTR served as the control.Dual luciferase reporter assay(All data are expressed as the mean ± SD ,kP < 0.05,lP < 0.01,miR-224-5p + TXNIP WT 3′-UTR vs miR-224-negative + 3’-UTR-mutant groups;n = 3);C:Neonatal mouse CMs subjected to hypoxia were pre-treated with EVs derived from ADSCs(ADSCs group),ADSCs overexpressing miR-224(ADSCs-AAV-miR-224 group),ADSCs overexpressing miR-224-NC(ADSCs-AAV-miR-224NC group)and ADSCs with miR-224 knocked-out(ADSCs-cas9-miR-224 group).Western blotting was used to detect the expression of hypoxia-inducible factor-1 alpha and TXNIP(n = 5);D:Neonatal mouse CMs subjected to hypoxia were pre-treated with EVs derived from ADSCs(ADSCs group),ADSCs overexpressing miR-224(ADSCs-AAV-miR-224 group),ADSCs overexpressing miR-224-NC(ADSCs-AAV-miR-224NC group)and ADSCs with miR-224 knocked-out(ADSCs-cas9-miR-224 group).Annexin V/PI assay was used to assess the degree of apoptosis in four groups(n = 5).All data are expressed as the mean ± SD,iP < 0.05,jP < 0.01,compared with CMs subjected to hypoxia were pre-treated with EVs derived from ADSCs group(ADSCs group).HP-EV:Hypoxia-preconditioned extracellular vesicles;NC-EV:Normoxic extracellular vesicle;ADSC:Adipose-derived mesenchymal stem cell;WT:Wildtype.
DlSCUSSlON
EVs are biocompatible,high-tissue penetrating,nano-sized secreted vesicles containing many types of biomolecules,including proteins,RNAs,DNAs,lipids,and metabolites.Their low immunogenicity and ability to functionally modify recipient cells by transferring diverse bioactive constituents make them an excellent candidate for a next-generation drug delivery system[20,28,29].Despite the tremendous achievements,clinical application of EVs remains challenging for the following reasons.There is no universally accepted gold standard for EVs extraction methods to meet clinical application,biosafety concerns regarding editing and modification of EVs,and the question of whether long-term clinical use of EVs could produce unacceptable side effects has not been resolved[30,31].Thus,the goal of this study was to improve the therapeutic efficacy of MSC-derived EVs against MI-induced CM death by using a safe hypoxia preconditioning methodin vitro,and to explore the mechanisms of the protective effects of EVs.
The idea of hypoxia preconditioning of ADSCs came from remote ischemic preconditioning(RIPC),which is a novel method where ischemia followed by reperfusion of one organ is believed to protect remote organs either due to release of biochemical messengers in the circulation or activation of nerve pathways,resulting in release of messengers that have a protective effect[32].With regard to the underlying mechanism of RIPC in cardioprotection,whether such preconditioning efficacy may extended applicationin vitroto provide a promising treatment by using EVs generated by ADSCs exposure to hypoxia preconditioning[33].For example,EVs generated by HP MSCs were adapted to improve traumatic spinal cord injuryviaits paracrine mechanisms and unfolded a myocardium preservation effect against ischemia-reperfusion injury[34,35].Thus,we proposed to explore whether anin vitroRIPC process can change contents of EVs derived from ADSCs to strengthen its intrinsical cardioprotective potential.Our results revealed that HP-EVs elicited more significant inhibiting effect of MI-triggered CM death than NC-EVs.Notably,HP-EVs were changed its contained miRNA expression(88 miRNA)after ADSCs exposure to preconditioning,10 of these upregulated miRNA are putative regulators of inflammasome activation based on the predicted binding affinity for TXNIP.We focused on the most upregulated miRNA(i.e.miR-224-5p)and verified both direct binding to TXNIP and a critical role for this interaction in the inhibition of MI-induced CM death.
Additionally,our results also objectively illustrated the fact that the timely opening of the infarction vessels and reducing the apoptosis of CMs during MI are equally crucial.Once delayed treatment occurs,CMs would change from a reversible injury state to necrosis,apoptotic necrosis,fibrous tissue replacement,and eventually to ventricular remodeling,which is often irreversible.Hence,for the treatment of MI,on the one hand,we should emphasize timely reperfusion therapy;on the other hand,we should preserve more CMs without irreversible injury,even death,until the moment of artery recanalization to maintain overall heart function after reperfusion therapy by enhancing hypoxia tolerance.In this study,HP-EVs exhibited a significant cardioprotective effect against aggravated apoptosis in MI caused by hypoxia and ischemia.Mechanistically,the benefit of CMs was mainly derived from effective hypoxia tolerance induced by HP-EVs.
The hypoxia tolerance of CMs induced by HP-EVs revealed that the HIF-1 transcription factor partly contributed to this benefit.HIF-1,of which the active subunit 1α undergoes proteasomal oxygendependent degradation,has an essential cardioprotective role and is a key mediator of the adaptability of the myocardium to hypoxia[36].Under aerobic conditions,hydroxylated HIF-1α is recognized by pVHL,which combines with HIF-1α in a ubiquitin ligase form to be exported into the cytoplasm,where HIF-1α is degraded.By contrast,hypoxia promotes the accumulation of unhydroxylated HIF-1α and translocation to the nucleus to initiate transcriptional activity[37].The role of HIF-1 in ischemic cardiomyopathy is different from its role in mediating oxygen homeostasis,inflammation,autoimmunity,and tumor metastasis under hypoxic conditions[38].In MI,hypoxia and slight mROS generation induced by the unstable membrane potential of mitochondria,which are two independent factors that increase TXNIP expression in CMs,expedite the export and degradation of HIF-1α through the pro-oxidative stress function of TXNIP[39,40].Considering that CMs contain a large number of mitochondria,once ischemia and hypoxia occur,mitochondria dysfunction induces the high expression of TXNIP and high level of HIF-1α degradation,which decreases the tolerance of hypoxia of CMs,accelerating their death.Clinically,if the duration of the MI exceeds its 12 h-therapeutic time window or time from first medical contact to re-opening blocked blood vessels,which was defined as “90 min doorto-balloon time”,CMs would enter an irreversible process of death,when even reperfusion therapy did not help[40,41].It is assumed that reperfusion therapy can be performed in a fixed time period;thus,therapies that facilitate CM survival from onset of MI to reperfusion therapy(elevating the hypoxia tolerance of CMs in this fixed time period)are particularly significant.Moreover,when reperfusion/reoxygenation therapy is delayed or prolonged,enhancing the hypoxia tolerance of CMs may be an ideal choice in order to preserve more CMs that cannot regenerate rather than waiting for irreversible injury to occur before treatment.Thus,increasing hypoxia tolerance and timely reperfusion therapy appear to be ‘two-horse carriages’ in preserving CMs in the early stage of MI.
As a core transcription factor antagonizing apoptosis and inflammation and promoting proliferation and hypoxia tolerance under hypoxia conditions,HIF-1α is key for controlling the expression of a myriad of genes involved in the hypoxic response;thus,its role in ischemic cardiomyopathy has received increasing attention[42].In our previous study,we revealed that HP-EVs primarily target TXNIP to alleviate myocardial IR injury post-reperfusion therapy[43].Theoretically and mechanistically,we demonstrated that TXNIP,which interacts with HIF-1α,involves the degradation of HIF1α through accelerating the nuclear export of ubiquitinated HIF-1αviathe CRM-1 nuclear export pathway in hypoxic conditions.Thus,HIF-1α-induced tolerance to hypoxia is weakened under hypoxic conditions to exert severe ischemia or hypoxia injury in CMs,unlike its effect on tumor metabolism and angiogenesis under hypoxic conditions.HIF-1α has long been mainly considered an oxygen homeostasis regulator,probably because of its characteristic transcriptional activation[44,45].However,recent studies have indicated that HIF-1α contributes to anti-apoptosis,regulation of energy metabolism,and collateral vessel generation[46,47].Therefore,considering that HIF-1α has the potential to regulate hypoxia-related injury and our previous studies on the inhibitory effects of HP-EV on TXNIP,we confirmed that highly expressed TXNIP in hypoxic conditions triggers the degradation of HIF-1α.Furthermore,by targeting TXNIP,HP-EVs improve the prognosis of MI.
The limitation of this study was,due to the difficulty in effectively inhibiting the action of the myocardial proteasome,we did not assess HIF-1α ubiquitinationin vivo.The interaction among pVHL,TXNIP,and HIF-1α as well as the mechanism of the CRM-1 nuclear export pathway under hypoxia/MI conditions remains to be further studied.
CONCLUSlON
In conclusion,our study demonstrated that EVs generated by ADSCs subjected to hypoxia preconditioning showed more significant cardioprotection against MI than EVs derived from normoxic ADSCs,partly due to the abundance of miRNAs targeting TXNIP in HP-EVs.TXNIP-aggravated ubiquitination of HIF-1α in CMs exposed to MI determines the tolerance of cells to hypoxia(Figure 9).Therefore,we propose that the downregulation of TXNIP by EV-associated miRNAs prevents the nuclear export and ubiquitination of HIF-1α,which protect CMs against early-stage ischemic injury by sustaining the transcriptional activity of HIF-1α.Our study provides novel insights into therapeutic approaches and the pathogenesis of MI and reveals that EVs derived from HP MSCs could help improve myocardial hypoxia tolerance when applied in the early stage of MI.
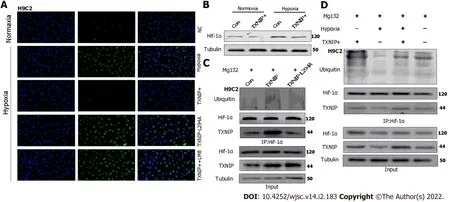
Figure 8 Thioredoxin-interacting protein promotes the ubiquitination of hypoxia-inducible factor-1 alpha through the chromosomal region maintenance-1 nuclear export pathway.A:H9C2 cardiomyocytes(CMs)overexpressing thioredoxin-interacting protein(TXNIP)or TXNIP-L294A mutant as indicated and leptomycin B were used to inhibit the chromosomal region maintenance 1 nuclear export pathway.The expression and cellular localization of hypoxia-inducible factor-1 alpha(HIF-1α)was analyzed by immunofluorescence staining;B:H9C2CMs were treated with hypoxia or normoxia and TXNIP was overexpressed as indicated.The expression of HIF-1α was analyzed by western blotting;C:H9C2 CMs were pretreated with MG132 for 2 h and TXNIP or TXNIPL294A-mutant were overexpressed as indicated.The interaction of TXNIP and HIF-1α,and HIF-1α ubiquitination were assessed by immunoprecipitation with an anti-HIF-1α antibody;D:H9C2 CMs were pretreated with MG132 for 2 h.TXNIP was overexpressed and hypoxia treatment was applied as indicated.The interaction of TXNIP and HIF-1α,and HIF-1α ubiquitination were assessed by immunoprecipitation with an anti-HIF-1α antibody.
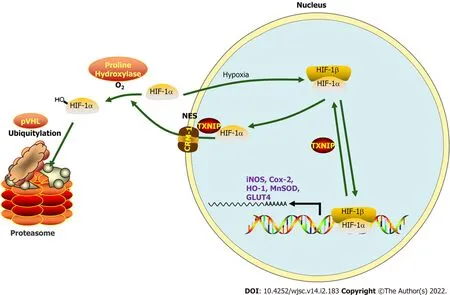
Figure 9 During myocardial infarction or hypoxia,hypoxia-inducible factor-1 alpha export is chromosomal region maintenance-1-dependent in the presence of thioredoxin-interacting protein,based on the association of thioredoxin-interacting protein with chromosomal region maintenance-1.Thioredoxin-interacting protein(TXNIP)-induced hypoxia-inducible factor-1 alpha(HIF-1α)nuclear export may be hypoxia-independent,which triggers ubiquitination and degradation by the proteasome in cardiomyocytes.As a transcription factor,the function of HIF-1 in promoting targeted gene transcription is abolished by hypoxia/myocardial infarction-triggered TXNIP activation.Our research aimed to clarify this process by hypoxiapreconditioned extracellular vesicles and the underlying mechanism.
ARTlCLE HlGHLlGHTS
Research background
Previous studies have demonstrated that extracellular vesicles(EVs)derived from mesenchymal stem cells(MSCs)reveal the cardioprotective effects against myocardial infarction(MI).Hypoxia-preconditioned EVs(HP-EVs)derived from MSCs are thought to have better cardioprotective effects,and the underlying mechanisms have garnered increasing attention from scholars.
Research motivation
Although some scholars have focused on the effect of hypoxia preconditioning on MSCs,the underlying mechanisms remain unclear.Thus,this study focused on the mechanism underlying the cardioprotective effect of HP-EVs from MSCs.
Research objectives
We explored the cardioprotective mechanism of HP-EVs from MSCs.
Research methods
HP-EVs from mouse adipose-derived MSCs(ADSCs)were extracted,and their cardioprotective effect on improving the survival of cardiomyocytes(CMs)and ameliorating cardiac function were evaluated by Evans Blue/2,3,5-triphenyltetrazolium chloride staining and echocardiography.Mechanistically,microRNA(miRNA)sequencing was adopted to investigate the functional RNA diversity between HPEVs or normoxic EVs(NC-EVs)from mouse ADSCs.Subsequently,the molecular mechanism of EVs in mediating thioredoxin-interacting protein(TXNIP)and TXNIP-mediated hypoxia-inducible factor-1 alpha(HIF-1α)ubiquitination were verified by the dual-luciferase reporter assay,immunoprecipitation,western blotting,and immunofluorescence.
Research results
HP-EVs reduced both infarct size(necrosis area)and the degree of apoptosis to a greater extent than NC-EVs in CMs subjected to hypoxia in vitro and mice suffering from MI in vivo.We showed that EV miRNA224-5p directly bound to the 3’-untranslated region of TXNIP and had a critical protective role against hypoxia-associated CM injury.Our results suggested that MI triggered TXNIP-mediated HIF-1α ubiquitination and degradation via the chromosomal region maintenance 1-dependent nuclear transport pathway in CMs,which led to aggravated injury and hypoxia tolerance in CMs in the early stage of MI.
Research conclusions
The anti-apoptotic effect of HP-EVs,which improves tolerance toward MI or hypoxic conditions and alleviates the degree of CM apoptosis until reperfusion therapy,may partly result from EV miR-224-5p targeting TXNIP.
Research perspectives
This study partly reveals the mechanism underlying the cardioprotective effect of HP-EVs and provides insights into potential therapies against MI.
FOOTNOTES
Author contributions:Mao CY,Zhang TT,Li DJ,and Zhou E contributed equally to this work,performed the experiments,and reviewed and edited the manuscript;Fan YQ and He Q wrote the paper;Zhang JF,and Wang CQ conceived of and funded the study;all authors read and approved the final manuscript.
Supported byNational Natural Science Foundation of China,No.81870264 and No.81470546;the Shanghai Committee of Science and Technology,No.18411950500;the Major Disease Joint Project of Shanghai Health System,No.2014ZYJB0501;and Talent Cultivation Project of The Ninth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine,No.JC202005.
lnstitutional review board statement:The study was reviewed and approved by the Institutional Review Board of Shanghai Ninth People’s Hospital.
lnstitutional animal care and use committee statement:All animal procedures were approved by the Shanghai Ninth People’s Hospital Institutional Ethics Committee and conducted in accordance with the guidelines of the Directive 2010/63/EU of the European Parliament.
Conflict-of-interest statement:The authors declare that they have no competing interests regarding this study.
Data sharing statement:The data that support the findings of this study are available from the corresponding author upon reasonable request.
ARRlVE guidelines statement:The authors have read the ARRIVE guidelines,and the manuscript was prepared and revised according to the ARRIVE guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial(CC BYNC 4.0)license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is noncommercial.See:https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Cheng-Yu Mao 0000-0001-9740-8835;Tian-Tian Zhang 0000-0001-8218-2757;Dong-Jiu Li 0000-0003-4372-6613;En Zhou 0000-0002-6073-8037;Yu-Qi Fan 0000-0002-0084-2895;Qing He 0000-0002-1078-350X;Chang-Qian Wang 0000-0002-7611-7761;Jun-Feng Zhang 0000-0001-9530-3263.
S-Editor:Fan JR
L-Editor:A
P-Editor:Zhang YL
杂志排行

World Journal of Stem Cells的其它文章
- Abnormal lipid synthesis as a therapeutic target for cancer stem cells
- Transcription regulators differentiate mesenchymal stem cells into chondroprogenitors,and their in vivo implantation regenerated the intervertebral disc degeneration
- Anti-fibrotic effect of adipose-derived stem cells on fibrotic scars
- Physical energy-based ultrasound shifts M1 macrophage differentiation towards M2 state