以突眼为主要症状的朗格汉斯细胞组织细胞增生症1例
2022-05-31陈仕胜吴建洺高宇
陈仕胜,吴建洺,高宇
温州医科大学附属第二医院育英儿童医院 皮肤科,浙江 温州 325027
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis, LCH)认为是由丝裂原活化蛋白激酶通路突变引起的骨髓前体细胞炎症性肿 瘤[1],临床表现及预后差异较大,轻者仅单器官受累,部分自行缓解,重者多系统受累,较难治疗,病死率高达20%[2]。早期轻微的临床表现应考虑到该病的可能,及时评估其他系统的累及对于早期诊断和治疗至关重要。现报告1例以突眼为主要症状的LCH。
1 病例资料
患儿,男,3岁,因右侧突眼伴有泪液增多5个月就诊。患者5个月余前无明显诱因出现右侧眼球微突伴有泪液增多,低热,体温波动于38~38.5 ℃之间,至当地医院行退热处理(具体不详),热退后突眼症状有所缓解。但此后突眼症状不断加重,患者前往温州医科大学附属眼视光医院就诊,行眼眶CT平扫及眼部B超检查,考虑“眼部囊肿”,建议至温州医科大学附属第二医院育英儿童医院行全身检查,遂于2019年5月1日入住我院血液科。
入院查体:意识清,全身皮肤未见黄染,右侧眼球突出明显,躯干部可见散在米粒大小淡红色丘疹,质软,互不融合,头顶处可见油腻性厚痂(见图1)。双肺呼吸音稍粗,心率100次/min,律齐,腹软,肝脾肋下可触及轻度肿大,无触痛,肠鸣音活跃,病理征阴性。实验室检查:血常规:C反应蛋白48.39 mg/L,白细胞11.85×109/L,血小板527×109/L;尿常规:白细胞计数22.16/HP,尿蛋白定性阳性(2+);血沉:103 mm/h;白细胞介素-6: 73.99 pg/mL。特殊检查:头部螺旋CT平扫:右侧额骨、双侧上颌窦各壁、双侧蝶骨及蝶骨斜坡、C2椎体及附件呈虫蚀状多发骨质破坏,周围见多发软组织肿块形成(见图2)。胸腹部皮疹病理活检:表皮真皮交界处可见组织细胞及少量淋巴细胞浸润。免疫组化:CAM5.2(-),CD1a(+),CD45(+),CD68(+),Ki-67(约30%+),S-100(+),SOX-10(-),见图3。
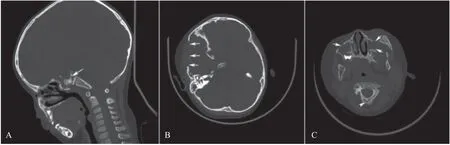
图2 患儿入院时头部螺旋CT平扫(白色箭头所指为病灶)
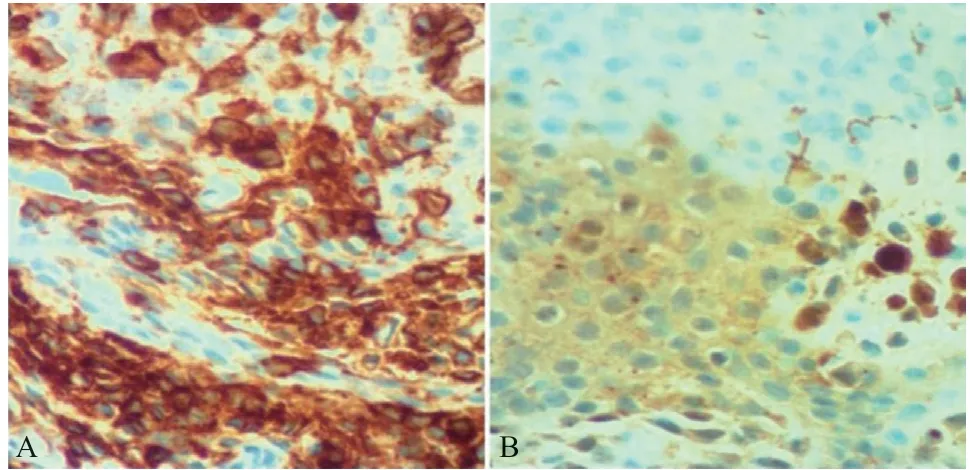
图3 胸腹部皮损活检组织的免疫组化染色结果(×100)
诊断:多系统型朗格汉斯细胞组织细胞增生症(MS-LCH)。
治疗:2019年5月21日开始采用长春地辛联合泼尼松化疗方案(即VP方案)治疗。经6次VP方案化疗后,MRI评估患儿病灶无明显缩小,眼球突出无改善,遂赴上海交通大学医学院附属新华医院儿童血液肿瘤科,于2019年8月30日至2019年10月14日行Group2化疗方案治疗。2019年11月4日行头颅MRI复查,评估病情处于中间状态。2019年11月12日开始,予3轮难治性LCH方案即PV×6周+VP16化疗方案治疗,再次复查头颅MRI,病灶较前稍好转,予行甲氨蝶呤联合6-巯基嘌呤3周联合VP方案1周,序贯维持治疗方案治疗。目前随访中,2022年3月1日最后1次电话随访,患儿一般情况可,右眼球突出较前未见加重。
2 讨论
LCH是一组以CD1a+/Langerin+朗格汉斯细胞在组织中的异常积累和广泛的器官受累为特征的疾病,以前被称为组织细胞增生症X,1987年组织细胞学会工作组建议统一命名为LCH[3]。流行病学研究报道LCH的发生率为3~5/百万;大多数患者是3岁以下的儿童,成人的发病率约为2/百万。北欧国家的高加索人的发病率似乎较高,而亚洲和非洲的发病率较低[4]。
LCH的发病机制不明。2010年,有研究报道从超过一半的LCH患者样本中发现了BRAF(V600E)的功能获得性突变,这使得人们对LCH发病机制的理解取得了新突破[1]。随后有研究报告称,所有的LCH病例存在ERK磷酸化,表明LCH很可能是一种克隆性的髓系肿瘤[5]。因此LCH在2016年组织细胞学会分类中被修订定义为炎性髓系肿瘤[3]。研究进一步显示在LCH累及多系统的患者中BRAF(V600E)频率更高。除了V600E突变,研究还发现存在V600D、BRAFG466R、MAP2K1等MAPK(RAS-RAF-MEK-ERK)通路上其他基因的突变[6]。这些新的发现,能极大地提高LCH的早期诊断率,如通过液态活检检测循环血中是否存在突变的细胞,可以提早发现那些累及不易进行活检的器官和组织的LCH。更重要的是,可以针对相应的突变开发小分子抑制剂,实现精准治疗[7]。
LCH诊断需结合临床表现、影像学检查和组织病理结果综合判断。临床表现有显著差异,从单个系统受累,到难治性多系统受累。三分之二的儿童患者是单系统累及,最常见的是骨骼,还有皮肤或淋巴结。大约25%的病例累及皮肤[8]。既往人们习惯根据累及部位不同将LCH分4种:①骨嗜酸性肉芽肿,临床表现为骨骼损害引起相应表现;②Hand-Schuller-Christian病,以突眼、颅骨损害和尿崩症为主要症状;③Letterere-Siwe病,累及多系统,如骨、皮肤、肝、脾脏和淋巴结等部位;④Hashimotoe Pritzker病,即先天性自愈性组织细胞增生症[2]。有些具有重叠表现的病例不能完全适合此分类。本例有显著的突眼症状和颅骨损害,但无尿崩症状,归类为Hand-Schuller-Christian病也不合适。LCH目前按受累系统及程度分为:单系统型LCH(SS-LCH)和多系统型LCH(MS-LCH)。SS-LCH指仅侵犯单个系统,又分单灶型和多灶型;MS-LCH是指两个以上器官累及,分为A型(无功能损害)和B型(有功能损害)。这样的分类能反映临床转归、治疗策略的选择及预后。单个低风险器官累及预后良好,一般能自然缓解或者治愈,多系统累及伴风险器官(肝脏、脾脏或者造血系统)累及需要联合化疗,才能提高长期生存率[9]。本例患儿以突眼症状就诊,皮疹为非特异性湿疹样表现,及时进行皮损组织病理活检及免疫组织病理检查,发现CD1a(+),S-100(+),结合临床表现及影像学多发骨损害及多脏器受累,诊断MS-LCH成立。需与其他组织细胞增多症如非朗格汉斯细胞组织细胞增多症、恶性组织细胞增生症等鉴别[10],病灶组织的病理活检免疫组化检查有助于这类疾病的鉴别诊断。
综上,LCH临床少见,临床表现异质性明显,需要临床医师提高警惕,对于一些不典型症状的鉴别诊断需考虑到该病。诊断分类需要结合受累器官的多少及是否存在风险器官受累。治疗根据诊断分类针对性治疗,单个低风险器官累及可以随访观察,多系统累及或者风险器官累及需要联合化疗。
