MLKL基因敲除对猪伪狂犬病病毒复制的影响
2022-05-31谢思豪勾红潮卞志标蔡汝健臧莹安李春玲
谢思豪,勾红潮,卞志标,李 斌,蔡汝健,臧莹安,李春玲
(1.仲恺农业工程学院,广州 510225;2.广东省农业科学院动物卫生研究所,广东省畜禽疫病防治研究重点实验室,农业农村部兽用药物与诊断技术广东科学观测实验站,广州 510640;3.广州市从化区动物卫生监督所,广州 510900)
CRISPR/Cas9基因编辑技术是一种简单、可靠的基因组编辑工具[1-2],只需针对目的基因序列设计特异性的sgRNA,通过sgRNA对目的基因核苷酸序列进行识别定位,引导Cas9蛋白特应性结合并发挥核酸内切酶活性,造成特定位点上DNA双链损伤,其在修复的过程中可实现基因组特定位点的DNA缺失、插入或碱基突变[3]。目前,CRISPR/Cas9基因编辑技术已广泛用于各种细胞的基因编辑和修饰,为疫苗生产过程中提高病毒产量提供了一种可行性策略。张爽等[4]利用CRISPR/Cas9基因编辑技术构建了干扰素调节因子3(interferon regulatory factor 3,IRF3)基因敲除PK-15细胞系,与对照细胞相比,敲除IRF3基因显著增加了猪伪狂犬病病毒(Pseudorabies virus,PRV)拷贝数。张越秀等[5]利用CRISPR/Cas9基因编辑技术构建了敲除2’,5’寡腺苷酸合成酶2(OAS2)基因的猪肾上皮细胞(porcine kidney epithelial cells,PK-15),将猪瘟病毒(Classical swine fever virus,CSFV)感染PK-OAS2-KO细胞系,结果显示敲除OAS2基因能够显著促进CSFV复制。
PRV属于疱疹病毒科α-疱疹病毒亚科,具有143 kb的双链线性DNA,编码70多种蛋白质[6]。PRV的自然宿主是猪,但它可以感染大多数哺乳动物,包括猪、牛、马、啮齿动物和犬,对养殖业危害巨大[7-8]。 近年来人感染PRV的情况多有发生[9]。 PRV在猪外周神经系统的三叉神经节中可建立终身潜伏感染[10]。在某些情况下,PRV可重新激活,继而导致PRV在猪场的反复流行,难以控制和根除。目前,猪伪狂犬病的防控以疫苗接种为重要手段,其中以PRV Bartha-K61为代表[11]。因此,提高PRV Bartha-K61病毒产量对疫苗生产具有重要意义。
程序性细胞死亡是哺乳动物宿主防御病原感染的重要途径,坏死作为程序性细胞死亡的一种炎症形式,在对抗病毒感染中起着重要作用[12]。坏死的激活依赖于受体相互作用蛋白激酶3(receptor-interacting protein kinase 3,RIPK3)和混合谱系激酶结构域样(mixed lineage kinase domain-like,MLKL)的信号级联反应[13-14]。MLKL是RIPK3的功能底物,即在细胞坏死中被RIPK3激活的下游蛋白[15]。在MLKL磷酸化被激活后形成寡聚体,这些寡聚体被转移到细胞质和细胞内膜上,导致细胞坏死[16-17]。前期研究表明,稳定敲低RIPK3或MLKL基因表达可以减少PRV GD-WH野毒变异株诱导的细胞坏死,并可以提高PRV GD-WH野毒变异株病毒滴度[18]。为了构建可促进PRV Bartha-K61增殖的细胞株,在前期研究基础上,本研究利用CRISPR/Cas9基因编辑技术对PK-15细胞MLKL基因对应的染色体序列进行双sgRNA剪切,通过单克隆纯化获得MLKL基因缺失的PK-15细胞株(PK-15 MLKL-KO),以期为提高PRV Bartha-K61等疫苗株的培养滴度提供新的策略。
1 材料与方法
1.1 细胞、病毒与质粒
PK-15细胞、PRV GD-WH株(PRV GD-WH)、PRV Bartha-K61疫苗株(PRV Bartha-K61)均由广东省农业科学院动物卫生研究所猪病研究室保存;大肠杆菌Trans10感受态细胞购自全式金生物技术有限公司;CRISPR/Cas9载体质粒pX459 pSpCas9-2Apuro-MCS和辅助载体质粒EZ-GuideXH均购自Addgene公司。
1.2 主要试剂
限制性核酸内切酶BbsⅠ、Hind Ⅲ、XhoⅠ均购自New England Biolabs公司;T4 DNA Ligase、T4 DNA Ligase Buffer均购自TaKaRa公司;Lipofectamine 3000转染试剂购自Thermo Fisher Scientific公司;碘化丙啶(PI)溶液购自Beyotime Biotechnology公司;辣根过氧化物酶(HRP)标记的山羊抗兔IgG多克隆抗体、HRP标记的山羊抗小鼠IgG多克隆抗体、兔抗MLKL多克隆抗体、小鼠抗GAPH单克隆抗体均购自Beyotime Biotechnology公司。
1.3 引物设计及合成
根据NCBI数据库公布的猪MLKL基因序列(Gene ID:100736836),通过在线CRISPR设计工具(http:∥crispr.mit.edu/)设计2对特异性sgRNA序列(表1)。引物均由生工生物工程(上海)股份有限公司合成。

表1 MLKL基因sgRNA序列
1.4 MLKL-sgRNA载体构建
将合成后的单链sgRNA退火形成双链,分别与经BbsⅠ酶切的CRISPR/Cas9载体pX459 pSpCas9-2A-puro-MCS和辅助载体EZ-GuideXH通过T4 DNA Ligase 16 ℃连接过夜,获得pX459-sgRNA1和EZ-sgRNA2重组质粒并测序鉴定。以Hind Ⅲ和XhoⅠ对获得的2个重组质粒进行双酶切,回收后将线性化酶切产物16 ℃连接过夜,转化大肠杆菌Trans10感受态细胞,通过氨苄抗性平板筛选挑取单克隆,进行菌落PCR筛选,将筛选到的阳性克隆质粒进一步测序验证。
1.5 PK-15细胞培养与转染
将PK-15细胞培养于含10%胎牛血清和1%青霉素-链霉素的DMEM培养基中,置于37 ℃、5% CO2培养箱中培养。转染前将生长状态良好的PK-15细胞接种至6孔板中培养,当细胞汇合度达到70%~80%时,按照Lipofectamine 3000转染试剂说明书将5 μg MLKL-sgRNA载体转染至PK-15细胞中。
1.6 敲除MLKL基因的单克隆细胞株筛选
细胞转染24 h后,更换含有0.70 μg/mL嘌呤霉素的10%胎牛血清和1%青霉素-链霉素的DMEM培养基中进行药物筛选,连续筛选5 d,观察到阴性对照组细胞全部死亡后,将得到的阳性细胞继续培养1周后消化成单个细胞,使用有限稀释法将药物筛选得到的阳性细胞稀释至96孔板中继续培养,约2周后挑选生长状态良好的单克隆细胞进行鉴定。
1.7 MLKL基因敲除PK-15细胞株的鉴定
从96孔板中挑选生长状态良好的单克隆细胞进行扩大培养,连续传代到P10代后通过PCR、测序和Western blotting对MLKL基因的稳定敲除效果进行鉴定。取部分细胞提取DNA,使用针对敲除靶点设计的特异性引物进行PCR扩增,引物序列为:F:5′-GCCATCTCTTACCTCCCCTGA-3′;R:5′-AAACTAAGGCTGGAAGGGAGCA-3′,退火温度58 ℃,预计扩增片段长度为1 149 bp。PCR扩增产物经琼脂糖凝胶电泳检测后,送至生工生物工程(上海)股份有限公司进行测序,检测碱基插入或者缺失情况。通过Western blotting检测MLKL蛋白的表达,筛选MLKL基因敲除的PK-15 MLKL-KO细胞株。
1.8 病毒滴度测定
将PK-15和PK-15 MLKL-KO细胞接种在6孔板中,待细胞汇合度至70%~80%时,以MOI=10的PRV GD-WH和PRV Bartha-K61分别感染PK-15和PK-15 MLKL-KO细胞,并置于37 ℃、5% CO2培养箱吸附1 h。吸附结束后弃去接种物,用PBS清洗细胞3次,更换为维持培养基进行培养。按照12、24及36 h不同时间点收取细胞上清病毒液,将病毒液反复冻融3次,离心取上清液,将上清液分别10倍递减梯度稀释,感染96孔板中的PK-15细胞,病毒感染后共观察4 d,记录各孔的病变情况,按照Reed-Muench法测定各上清液的病毒滴度。
1.9 细胞坏死测定
将PK-15和PK-15 MLKL-KO细胞接种在96孔板中,待细胞汇合度至70%~80%时,以MOI=10的PRV GD-WH和PRV Bartha-K61分别感染PK-15和PK-15 MLKL-KO细胞,36 h后弃细胞培养基,用PI溶液在4 ℃下染色10 min,于荧光显微镜下观察细胞的PI染色情况。
1.10 统计学分析
所有试验至少重复3次,使用GraphPad Prism 6.0软件进行数据分析,结果以平均值±标准差表示,以P<0.05为差异显著性判断标准。
2 结 果
2.1 重组质粒MLKL-sgRNA构建
pX459-sgRNA1和EZ-sgRNA2重组质粒经Hind Ⅲ和XhoⅠ双酶切(图1A),酶切产物连接、转化,将筛选到的阳性克隆质粒进一步测序验证,测序结果表明MLKL-sgRNA载体构建成功(图1B)。
2.2 MLKL基因敲除PK-15单克隆细胞系构建
琼脂糖凝胶电泳检测结果显示,出现1 149和502 bp 2条条带(图2A);对2条条带进行回收、纯化和测序,序列比对分析结果显示,PK-15 MLKL-KO细胞在sgRNA靶点处缺失647 bp(图2B)。通过Western blotting对PK-15 MLKL-KO细胞中MLKL基因敲除效果进一步鉴定,结果显示,PK-15 MLKL-KO细胞未检测到MLKL蛋白的表达(图2C)。表明本试验成功构建了MLKL基因敲除的PK-15 MLKL-KO细胞株。
2.3 MLKL基因敲除对PRV GD-WH和PRV Bartha-K61病毒滴度的影响
由图3A可知,感染后12~24 h,PRV GD-WH在PK-15 MLKL-KO细胞中的病毒滴度极显著或显著高于PK-15细胞(P<0.01;P<0.05);感染后36 h,PRV GD-WH在PK-15和PK-15 MLKL-KO细胞中的病毒滴度无显著差异(P>0.05)。由图3B可知,感染后12~36 h,PRV Bartha-K61在PK-15 MLKL-KO细胞中的病毒滴度极显著高于PK-15细胞(P<0.01),其中,PRV Bartha-K61感染PK-15和PK-15 MLKL-KO细胞24 h后,病毒滴度分别为105.1和106.3TCID50/mL,提高了约15.8倍;在感染36 h后,病毒滴度分别为105.9和106.9TCID50/mL,提高了10倍。
2.4 MLKL基因敲除对PRV GD-WH和PRV Bartha-K61诱导细胞坏死的影响
PRV GD-WH和PRV Bartha-K61感染PK-15和PK-15 MLKL-KO细胞后,于荧光显微镜下观察细胞的PI染色(红色荧光)的情况,结果显示,PK-15 MLKL-KO细胞感染组PI阳性细胞明显少于PK-15细胞感染组(图4)。

①A,pX459-sgRNA1和EZ-sgRNA2重组质粒双酶切产物;B,MLKL-sgRNA测序结果。②M,DL5000 DNA Marker;1,EZ-sgRNA2;2,pX459-sgRNA1①A,Double digestion products of pX459-sgRNA1 and EZ-sgRNA2 recombinant plasmids;B,Sequencing results of MLKL-sgRNA.②M,DL5000 DNA Marker;1,EZ-sgRNA2;2,pX459-sgRNA1图1 MLKL基因敲除重组质粒的构建Fig.1 Construction of MLKL gene knockout recombinant plasmid
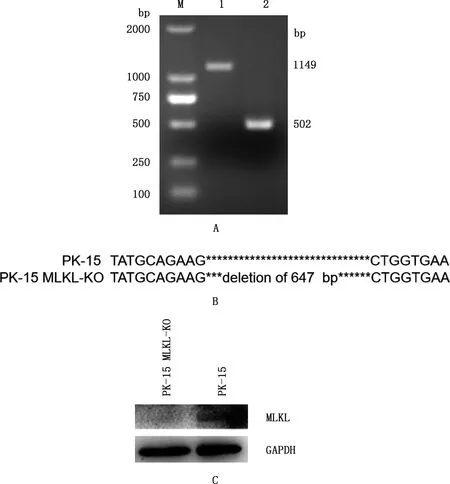
①A,MLKL基因敲除PK-15细胞PCR鉴定;B,敲除MLKL基因的PK-15细胞测序结果;C,PK-15细胞中MLKL蛋白的表达。②M,DL2000 DNA Marker;1,PK-15细胞;2,PK-15 MLKL-KO细胞①A,PCR identification of MLKL gene knockout PK-15 cell;B,Sequencing results of MLKL gene knockout PK-15 cell;C,Expression of MLKL protein in PK-15 cell.②M,DL2000 DNA Marker;1,PK-15 cell;2,PK-15 MLKL-KO cell图2 敲除MLKL基因的PK-15 细胞鉴定结果Fig.2 Identification results of PK-15 cells with MLKL gene knockout

*,差异显著(P<0.05);**,差异极显著(P<0.01);ns,差异不显著(P>0.05)*,Significant difference (P<0.05);**,Extremely significant difference (P<0.01);ns,No significant difference (P>0.05)图3 不同感染时间PRV GD-WH(A)和PRV Bartha-K61(B)病毒滴度Fig.3 Virus titer of PRV GD-WH (A) and PRV Bartha-K61 (B) at different infection time

图4 MLKL基因敲除对PRV GD-WH和PRV Bartha-K61诱导细胞坏死的影响(200×)Fig.4 Effects of MLKL gene knockout on PRV GD-WH and PRV Bartha-K61 induced cell necrosis (200×)
3 讨 论
CRISPR/Cas9基因编辑技术是近年来新发展起来的新型基因编辑技术,相较于与之前的ZFNs和TALENs基因编辑技术,CRISPR/Cas9基因编辑技术仅需要设计特异性sgRNA便可以对特定基因位点进行修饰,极大地降低应用成本、缩短时间[19]。同时,CRISPR/Cas9基因编辑技术具有特异性高、脱靶率低且可以同时对多个位点进行编辑的优势。为了提高编辑的成功率,本研究使用了双sgRNA编辑系统,经鉴定建立的PK-15 MLKL-KO细胞株成功敲除了MLKL基因647 bp,Western blotting也未检测到MLKL蛋白表达。
在减毒活疫苗的生产过程中,提高病毒滴度是关键。CRISPR/Cas9介导的RNaseL敲除促进了PRV Bartha-K61疫苗株的复制[20]。为了提高Vero细胞中轮状病毒疫苗的低产量,研究者采用CRISPR/Cas9技术编辑了Vero细胞的NUE2、NAT9、COQ9、EMX2等基因,使轮状病毒疫苗的产量显著增加[21-23]。程序性细胞坏死是一种重要的宿主防御机制,通过诱导细胞死亡限制病毒增殖,在对抗病毒感染中起着重要作用。MLKL是程序性细胞坏死的关键执行者,脂多糖(LPS)、病毒核酸和干扰素等均可诱导MLKL所介导的细胞坏死[24-26]。本研究拟构建一个稳定敲除MLKL基因的PK-15细胞株,用于PRV Bartha-K61疫苗生产过程中提高病毒滴度,结果表明,敲除MLKL基因显著提高了PRV Bartha-K61的复制能力。与PK-15细胞相比,PRV Bartha-K61感染PK-15 MLKL-KO细胞24 h后,病毒滴度提高了约15.8倍;在感染36 h后,病毒滴度提高了10倍。此外,稳定敲除PK-15细胞MLKL基因可以提高PRV GD-WH病毒滴度,这与Gou等[18]研究结果一致。
程序性细胞坏死是重要的抗病毒宿主的防御,其通过防止受感染细胞成为病毒工厂来限制病毒的传播。 研究表明,Ⅰ型单纯疱疹病毒(HSV-1)[27]、牛痘病毒(VV)[28]、寨卡病毒(ZIKV)[29]、甲型流感病毒(IAV)[30]均能诱导程序性细胞坏死的发生。多种病毒感染模式的例子强调了程序性细胞坏死对宿主免疫的重要性。MLKL基因缺失促进了PRV GD-WH和PRV Bartha-K61在PK-15细胞中的复制,与PK-15细胞相比,PRV GD-WH和PRV Bartha-K61感染PK-15 MLKL-KO细胞后,坏死细胞明显减少。PK-15 MLKL-KO细胞对PRV Bartha-K61增殖的促进作用可能与程序性坏死通路的抑制和细胞活性的增加有关,而MLKL缺失细胞株可能有潜力用于生产其他病毒的减毒疫苗,但敲除MLKL基因影响PRV Bartha-K61增殖的分子机制仍需进一步深入研究。
4 结 论
本研究构建了MLKL基因敲除的PK-15细胞株,与PK-15细胞相比,PK-15 MLKL-KO细胞显著提高了PRV GD-WH和PRV Bartha-K61的复制和存活能力,PRV GD-WH和PRV Bartha-K61感染PK-15 MLKL-KO细胞后,坏死细胞明显减少。
