甲型流感病毒所致宿主细胞因子风暴的研究进展
2022-05-26嵇祝星刘晓文王晓泉刘秀梵
嵇祝星,刘晓文,王晓泉*,刘秀梵
(1.扬州大学农业部畜禽传染病学重点开放实验室,江苏 扬州 225000;2.江苏高校动物重要疫病与人兽共患病
防控协同创新中心,江苏 扬州 225000)
甲型流感病毒(Influenza A virus,IAV)是有囊膜的正粘病毒科成员,其基因组包含8 个节段的单股负链RNA[1]。IAV 是人呼吸道感染的重要病原,可以导致季节性的流行和大流行。回顾历史,共发生了4次流感大流行,分别是1918 年H1N1、1957 年H2N2、1968 年的H3N2 以及2009 年的H1N1 流感大流行[2]。至今,全球每年有10 亿左右人口会感染流感病毒,有294 000~518 000 人死于季节性流感[3],给人类健康及公共卫生安全带来巨大的威胁[4]。
IAV 感染宿主后,被天然免疫细胞的模式识别受体(PRRs)识别,触发下游级联信号,产生大量细胞因子。在这一过程中,各种细胞因子、趋化因子以及白细胞相互作用、交联,构成了一张复杂的细胞因子通讯网络,形成“细胞因子风暴”。细胞因子风暴从疾病的早期到后期的任一阶段均可能快速发生[5],它虽然能够抑制病毒复制,但也会使正常的肺组织细胞死亡或被破坏,从而造成肺损伤[6]。并且各种病因的脓毒症[7]、凝血障碍[8]和弥散性血管内凝血[9]也与细胞因子风暴密切相关。
据报道,H5N1 和H1N1 亚型流感病毒感染后会导致机体发生过度的细胞因子反应,产生细胞因子风暴[10]。值得注意的是,在季节性流行的轻度流感中很少观察到严重的细胞因子风暴,这表明细胞因子(趋化因子)水平与疾病的严重程度密切相关[11]。细胞因子单体及交叉调节作用对感染结果有重要意义,如宿主IL-1β、IL-6、TNF-α、IL-10、IP-10 等因子的分泌与流感的严重程度密切相关[12]。本综述阐述流感病毒感染后宿主细胞因子风暴的产生机制,并总结了一些具有代表性细胞因子之间的调控网络(图1)和在流感病毒感染后其在天然免疫反应中的分泌、调节过程及在其中发挥的作用。
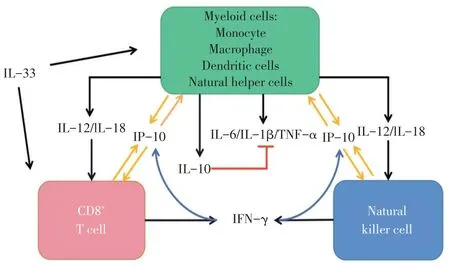
图1 甲型流感病毒诱导的“细胞因子风暴”中的主要细胞因子调控网络
1 白细胞介素-1 家族(Interleukin-1 family)
白细胞介素-1(IL-1)家族细胞因子包括11 个相关基因产物,据报道其中IL-1β、IL-18 和IL-33 与细胞因子综合征(Cytokine storm syndrome,CSS)相关。总之,IL-1 家族是一种促炎细胞因子,在流感病毒感染后诱导机体产生炎症反应并且向感染组织募集其他炎症细胞来清除病毒。
1.1 白细胞介素-1β(Interleukin-1β,IL-1β) H1N1亚型流感病毒感染人肺微血管内皮细胞和肺成纤维细胞后可诱导细胞表达多种细胞因子和趋化因子,而用IL-1β 受体拮抗剂或中和抗体阻断IL-1β 信号可以减轻流感引起的炎症[13],并且在感染初期和病毒复制高峰期阻断IL-1β 可显著减少宿主中性粒细胞向气道的募集[14]。且IL-1β 抗体可使AIV 感染小鼠的肺部炎症面积减少[15]。这些均说明IL-1β 是一种促炎细胞因子,阻断IL-1β 信号是治疗流感诱导炎症的一个潜在靶点。
在流感病毒感染过程中,IL-1β 的释放依赖于中性粒细胞介导的NOD 样受体热蛋白结构域相关蛋白3 型(NLRP3)炎症小体的激活[16]。最近一项实验也表明NLRP3 炎症小体的过度激活可通过IL-1β 介导的中性粒细胞募集来保护小鼠免受IAV 的感染[17]。值得注意的是,在IAV 感染的不同阶段,激活的NL⁃RP3 对宿主可能是保护性的,也可能是有害的。在流感病毒感染早期采用NLRP3 抑制剂阻断IL-1β 和IL-18 的成熟,可致小鼠发生致死性超敏反应,这表明IL-1β 在感染早期可以促进机体康复。但在感染后期,炎症小体诱导的IL-1β 和IL-18 的持续激活可能会破坏良性的炎症循环,从而导致较差的临床结果[18]。总之,病毒早期诱导的IL-1β 可以通过促进CD8+T 细胞的活性和抗体应答水平从而发挥保护作用,但如果整个应答过程中IL-1β 持续存在则会产生负面后果。这些研究也首次证明抑制NLRP3 是降低IAV 严重感染程度的新的治疗靶点。
1.2 白细胞介素-18(Interleukin-18,IL-18) IL-18是一种促炎细胞因子。在早期的研究中,IL-18 被当作IFN-γ 产生的诱导剂[19],但随着研究的深入,发现单独的IL-18 仅诱导机体产生少量的IFN-γ,而与IL-12/IL-15 的协同作用,可以诱导机体T 细胞产生高水平的IFN-γ[20-22]。
在流感病毒感染过程中,IL-18 的功能类似于IL-1β:其通过促进病毒特异性CD8+T 细胞产生的细胞因子和增强自然杀伤细胞的细胞毒性作用来改善流感病毒感染的严重程度[23]。在体外试验中,IL-18与I 型干扰素(IFN-I)协同可诱导机体T 细胞产生IFN-γ,并可通过多种机制促进病毒的清除[24]。
1.3 白细胞介素-33(Interleukin-33,IL-33) 与IL-1β 和IL-18 不同,IL-33 不刺激机体产生炎症小体。相反,它在内皮细胞和上皮细胞中以核蛋白的形式存在,并在炎性细胞死亡时释放。
最早由Goffic 等人发现,在IAV 感染过程中,不论是体内或是体外,IL-33 在小鼠肺部均有较高的表达水平[25]。之后的许多研究均表明IL-33 在病毒感染中对于宿主的保护作用:首先,它在诱导CD8+T 细胞产生IFN-γ 方面具有类似于IL-18 的作用[26];其次,IL-33 可以通过诱导先天淋巴细胞(ILC2s)产生双调蛋白(AREG)来促进肺组织的修复和稳态[27];除此以外,IL-33 是天然辅助细胞(Natural helper cells,NHC)产生Ⅱ型细胞因子的关键因子,它可以诱导肺NHC 产生大量其他细胞因子如:IL-4、IL-5 和IL-10[28]。
但最近有研究发现IL-33 可以抑制先天和适应性Th1 反应和细胞毒性反应,从而加重流感病毒诱导的急性炎症及哮喘症状。Lara 等认为IL-33/ST2 轴是控制流感诱导的哮喘症状的潜在药理学靶点,通过这一途径在患者的气道中进行局部短期治疗,可以降低IL-33 的分泌水平,并稳定哮喘的症状[29]。
2 白细胞介素-6(Interleukin-6,IL-6)
已有研究证明IL-6 可以在H1N1 亚型流感病毒感染时保护宿主,并且促进宿主中性粒细胞介导的病毒清除作用。IL-6 的缺失会致流感病毒在肺中持续存在,进而导致宿主明显的肺损伤甚至死亡[30]。另一项研究发现IL-6 通过减少成纤维细胞的堆积、促进上皮细胞的存活及增加巨噬细胞向肺内募集和增强巨噬细胞对病毒的吞噬作用,对流感所致肺损伤后的修复起着至关重要的作用[31]。这些均表明IL-6 在流感病毒感染后对宿主所起的保护作用,值得一提的是,流感病毒也能显著上调细胞因子信号转导抑制因子3(SOCS3)的表达来规避IL-6/STAT3 介导的免疫应答[32]。
3 白细胞介素-10(Interleukin-10,IL-10)
宿主免疫系统还利用多种抗炎细胞因子来抵抗过度的炎症反应。IL-10 是典型的抗炎细胞因子,它作用于淋巴样细胞以调节和限制其效应功能。IL-10的抗炎作用体现在抑制单核细胞、巨噬细胞产生致炎介质[33]。在流感病毒感染的过程中,阻断效应T 细胞产生IL-10 时会致宿主肺部炎症反应增强,从而导致宿主的致命性损伤[34]。有研究发现H1N1流感病毒感染后可以诱导强烈的IL-10和IL-6。其中IL-6可能是诱导IL-10 产生的介质[35],表明宿主可能通过IL-10来调节由于细胞因子风暴造成的损伤,这预示着IL-10 的表达量可以成为判断肺脏损伤程度的一个潜在标志。
4 肿瘤坏死因子-α(Tumor necrosis factor,TNF-α)
TNF-α 是流感病毒感染后产生的细胞因子风暴中典型的促炎细胞因子,TNF-α 主要由单核巨噬细胞及其他免疫细胞(包括DC 细胞、B 细胞、NK 细胞和T 细胞)产生。在H5N1 禽流感病毒(AIV)感染期间,I 型肿瘤坏死因子受体(TNFR1)缺陷小鼠的发病率显著降低,但在病毒复制方面其与野生型小鼠无差异[36]。这说明TNF-α 可能会加重流感症状的严重程度,但对病毒复制的影响有限。
然而,可溶性TNF-α(solTNF-α)可以通过TN⁃FR1 介导的免疫调节作用,抑制肺部炎症和炎性细胞的浸润,从而起到保护效果。流感病毒感染后,在solTNF-α 缺陷的小鼠中观察到细胞因子和趋化因子的过度表达和细胞浸润的增强,包括病毒特异性CD8+T 细胞反应的增强。在感染早期,若宿主缺少solTNF-α,逐渐增强的CD8+T 细胞反应会致宿主肺部的损伤加重[37]。与之相反,流感病毒会诱导机体上调TNFR2 的表达来降低机体产生solTNF-α 的能力[38]。这进一步提示TNFR2 的特异性拮抗可能有助于限制流感病毒感染后由TNF-α 引起的机体损伤。
5 IFN-γ(Interferon-γ,IFN-γ)
IFN-γ 是一种强大的抗病毒细胞因子。在病毒感染早期,主要由NK 细胞产生IFN-γ 来控制病毒的复制,且IL-12 有助于这一过程。在之后的免疫应答中,T细胞成为IFN-γ的主要来源。总体而言,IFN-γ对宿主具有保护作用:一项研究将H1N1 病毒感染体外分化的TC-1 细胞(主要产生IFN-γ 的CD8+T 细胞),结果表明TC-1 细胞产生的IFN-γ 可保护受体动物免受流感病毒的感染[39]。
虽然IFN-γ 的产生对病毒清除和适应性免疫反应的进展至关重要,然而作为细胞因子风暴的一部分,IFN-γ 的过度产生也会导致不良后果。最近一项研究指出:IFN-γ 在重症甲型H1N1 流感病毒感染所致的急性肺损伤中起重要作用, IFN-γ 单克隆抗体有望成为未来流感大流行的潜在治疗药物[40]。
6 趋化因子-10(Interferon-inducible protein-10,IP-10)
IP-10 是IFN-γ 所诱导产生的一类可以趋化淋巴细胞的CXC 趋化因子亚家族成员。IP-10 的表达水平与流感病毒感染后肺损伤的严重程度密切相关,并且在H1N1、H5N1 和H7N9 亚型流感病毒中均起重要作用。其中,在H7N9 亚型流感病毒感染后诱发的“细胞因子风暴”中,IP-10 表达水平的上调极为显著[41],这也意味着IP-10 可以作为流感病毒感染后肺部炎症水平的标志物[42]。
IP-10 也是参与IAV 诱导急性肺损伤的关键炎症因子:小鼠在注射IP-10 后再感染H1N1 亚型流感病毒,其肺部病变明显加重[43]。但是经IP-10 单克隆抗体治疗后,感染小鼠的细胞因子和趋化因子水平也明显低于同型对照感染小鼠。利用人IP-10 单克隆抗体治疗H1N1 亚型流感病毒PR8 株感染的小鼠,发现其也能显著改善该病毒感染引起的急性肺损伤[44]。表明IP-10 可能是预测流感病毒感染所致疾病严重程度的关键炎症因子,同时也可以作为治疗细胞因子风暴引起肺损伤的潜在靶点。
7 小结与展望
本文介绍了IAV 感染引起宿主的天然免疫细胞因子风暴:一方面其既对限制流感病毒感染起到积极作用:如诱导局部炎症、清除感染细胞、调节细胞和分子免疫反应、促进组织修复和动态平衡;另一方面,IL-33、TNF-α 等细胞因子可对宿主肺部造成严重的损害,如弥漫性肺泡损伤、纤维蛋白渗出等,最终会导致对机体免疫功能的抑制和全身脏器的功能障碍[45]。
本实验室在以往的研究中,也针对不同亚型AIV 诱导的细胞因子进行了检测。通过对不同亚型AIV 感染小鼠后采集的肺脏组织进行细胞因子蛋白水平、转录水平的检测,发现IL-4、IL-6、IL-10、TNF-α、IFN-γ、IP-10 等细胞因子均显著上调,并且病毒的致病性和对肺部损伤程度往往与其中一种或几种细胞因子的上调呈正相关。而且AIV 单点或多点氨基酸突变可能会增强病毒对小鼠的致病力:如H5N8 亚型流感病毒PB2 蛋白发生I283M 及K526R 联合突变后会造成宿主细胞因子风暴,损伤其脏器组织并促进细胞凋亡,从而利于病毒的扩散[46]。除此以外,本研究室还筛选了抗病毒药物,其中有些药物可以下调宿主细胞因子表达水平,增加小鼠的生存几率[47]。
除了关注不同亚型流感病毒与细胞因子的关系,也应注重病毒感染后机体拮抗病毒及细胞因子网络的内在联系,所以本研究室归纳了天然免疫细胞与主要细胞因子网络在对抗IAV 感染方面的作用,其中CD8+T 细胞、NK 细胞、巨噬细胞、单核细胞和中性粒细胞既是“生产者”,也是“效应器”,它们在分泌细胞因子和推动炎症的发展方面有着重要作用。其分泌的细胞因子各有分工,又相互联系。髓系细胞可以分泌IL-1 家族、IL-10、IL-12、IL-18等白细胞介素及TNF-α,IL-12 和IL-18 等可以促进CD8+T 细胞与NK 细胞分泌IFN-γ,而IL-10 可以反向调节IL-1 家族的分泌,这也是宿主的一种自我保护机制,即只能在一定程度上缓解细胞因子风暴的损伤程度。除此以外,趋化因子IP-10 也在该调控网络中起重要作用,它可以在IFN-γ 的诱导下由T、B淋巴细胞、NK 细胞、树突状细胞和巨噬细胞分泌。在分泌IFN-γ 后,IP-10 又可诱导各细胞的趋化、凋亡及抑制细胞生长。另外,IP-10 是流感病毒感染后诱导宿主急性肺损伤的关键炎症因子,也可作为炎症发展进程的指标之一。
随着对天然免疫过程中“细胞因子风暴”这一现象研究的深入,其在流感病毒感染过程中的作用也呈现出多元性与复杂性。本文详细描述了一些典型的细胞因子以及它们构成的互作网络,在后续研究中,本研究室还会关注其他保护性的细胞因子,包括诱导局部炎症和拮抗病毒的蛋白。有人将病毒感染直接诱导的细胞因子,如IFN-I、ISGs、AREG 归纳入“细胞因子风暴”的范围,这个观点也对[27,48]。因为其本质是由病原引起的宿主天然免疫反应的一种防御机制,从病毒识别到各种免疫蛋白的产生均是相互关联的动态过程,将它们联系起来更有利于了解“细胞因子风暴”这一生物学现象。另外,还应重点关注主要细胞因子的作用靶细胞及其与其他细胞因子之间的关系,如协同、竞争作用,这有利于深入探究“细胞因子网络”之间相互调控的机理。综上所述,本文总结了IAV 所致的细胞因子风暴中的重要因子,阐述了它们的特殊功能与动态联系,为深入了解“细胞因子风暴”的作用机制、探寻药物作用靶点提供了理论支持。
