形貌可控的CsPbBr3 钙钛矿纳米晶的制备及其形成动力学的原位光致发光研究*
2022-05-26陈雪莲巨博焦琥珀李燕钟玉洁
陈雪莲 巨博 焦琥珀 李燕 钟玉洁
(西安石油大学材料科学与工程学院,西安 710065)
通过添加不同比例的十二烷二酸(DDDA)配体,在室温下采用配体辅助再沉淀法制备CsPbBr3 钙钛矿纳米晶.利用X 射线衍射仪、透射电子显微镜、紫外分光光度计、荧光光谱仪等对纳米晶的相结构、微结构和光学性质进行了表征.结果显示:所有纳米晶产物均为立方相结构;而纳米晶形貌会随着DDDA 浓度的增加从纳米立方块逐渐转变为厚度均一的纳米片,同时光致发光光谱从绿光区移至蓝光区.结合实验室搭建的具有超高时间分辨率(约100 ms)的原位光致发光装置,实时监测了不同形貌纳米晶在形成过程中光致发光光谱的演变.结果表明:未使用DDDA 时前驱体经历了快速成核和尺寸分布聚焦生长后生成纳米立方块;DDDA的出现促进了纳米晶在形成早期生成各向异性的纳米片,然而随着反应时间的延长,具有不对称结构的纳米片会通过溶解-再结晶方式转变为热力学稳定的纳米立方块.本工作对形貌可控的钙钛矿纳米晶的精准合成具有一定参考价值.
1 引言
近几年,铯铅卤化物钙钛矿纳米晶(CsPbX3(X=Br,Cl,I) PNCs)成为研究者们关注的焦点材料,由于尺寸、形貌和组分的可调性而具有带隙可调[1,2]、高荧光量子产率[3]、窄荧光发射峰[4]、超高缺陷容忍度[5]等优异性质,在光伏、照明和新一代显示领域展现出极大的应用潜力[6−8].因此,制备出尺寸和形貌可控的CsPbX3PNCs 是获得高效稳定光电/光伏器件的前提.
目前,最常用的合成纳米晶的方法有高温热注射(HI)法[9]和配体辅助再沉淀(LARP)法[10].与HI 法相比,LARP 法在操作上更简易、不需要惰性气体保护,成本低,适合规模化合成,成为了制备CsPbX3PNCs 的一种重要方法.然而,这种方法的形成过程同样难以控制,得到的量子点产物往往存在结晶质量差、尺寸不均一和稳定性差等问题,导致了最终量子点器件低劣的性能和运行寿命,严重制约其商业化应用前景.大量研究表明,纳米晶为离子晶体,常用的表面配体如油胺和油酸并不能紧密地结合在纳米晶表面,而是一直处于高度脱吸附状态[11],通过选择合适的配体并调节配体的类型、浓度、链长及化学计量等,可以有效调控钙钛矿纳米晶的形貌和尺寸[12,13].例如,Sun 等[14]通过对有机酸和有机胺配体的链长进行改变,使纳米晶形貌出现零维的球形量子点、一维的纳米棒、二维的薄层纳米片和三维的纳米立方块.Pan 等[15]采用HI 法合成时,改变有机酸和有机胺配体链长,实现纳米晶形貌和尺寸的调控;当有机胺的链长越短生成的纳米片越薄,而降低有机酸的链长会导致纳米晶的尺寸增大.Shamsi 等[16]报道了通过改变反应时间、反应温度以及短链有机酸和有机胺配体的比例,可以调节纳米片的侧向尺寸最大可达1 µm.Bekenstein 等[17]和Akkerman 等[18]几乎同时报道了纳米片厚度调控的制备方法,研究发现纳米片的厚度逐渐降低时,表现出明显的量子尺寸效应,且荧光峰峰位逐渐发生蓝移.可见,通过对合成方法和工艺条件的经验性尝试和探索,对钙钛矿纳米晶的形貌和光电性能有一定改善,但难以实现高质量纳米晶的精细合成和性能优化.大量研究证实,建立微观机理与宏观参数及产物性能间的构效关系对提升材料的性能非常关键[19,20].然而,目前对钙钛矿纳米晶的形貌和尺寸调节的微观机理尚不清楚,最主要的障碍是钙钛矿纳米晶的形成过程非常快,常规手段难以捕捉到其形成动力学过程[21].
基于此,本文将通过十二烷二酸(dodecanedioic acid,C12H22O4,DDDA)与油胺、油酸配体共同使用(分子结构式如图1(a)所示),结合LARP方法成功制备出不同形貌的CsPbBr3PNCs.为了揭示纳米晶的形貌调控机理,基于荧光光谱仪搭建了具有高时间分辨能力的原位光致发光(PL)测试平台(如图1(b)所示),利用该平台仔细研究和讨论了纳米立方块和纳米片的形成动力学过程.

图1 (a) DDDA 的分子结构式;(b) 实验室搭建的原位光致发光(PL)装置图Fig.1.(a) Chemical structure of DDDA;(b) lab-based insitu photoluminescence (PL) device.
2 实验部分
2.1 药品与试剂
溴化铯(CsBr,99.9%)、溴化铅(PbBr2,99%)、十二烷二酸(DDDA,99%)、N,N-二甲基甲酰胺(DMF,GC 99.9%)和 油 胺(oleylamine,80%—90%)均采购于Aladdin 公司;油酸(oleic acid,90%)采购于Sigma-Aldrich 公司;甲苯、乙酸乙酯采购于国药集团化学试剂有限公司.
2.2 CsPbBr3 PNCs 的制备
首先制备所需前驱体.取10 mL 的DMF,放入50 mL 烧杯中,将其在一定温度下进行搅拌,称取0.4 mmol 的CsBr 和0.4 mmol 的PbBr2加入到搅拌的DMF 溶液中,随后加入0.5 mL 油胺和1 mL 的油酸作为稳定剂和表面活性剂,直至烧杯中的药品完全溶解后,停止搅拌.然后,将0.2 mL上述前驱体快速注入到剧烈搅拌的2 mL 甲苯溶液中.待反应10 s 后,溶液表现出了强烈的绿色荧光,停止搅拌,即制得全无机CsPbBr3纳米立方块.在保证上述实验条件不变的情况下,再将DDDA配体与PbBr2按照0.25∶1.00,0.5∶1.0,0.75∶1.00的摩尔比分别加入到前驱体溶液中,直至烧杯中的药品完全溶解后,停止搅拌.随后,将0.2 mL 上述前驱体快速注入到剧烈搅拌的2 mL 甲苯溶液中,待反应20 s 后,溶液呈现出蓝绿混合光或蓝光,即制得纳米立方块与纳米片的混合或纯的纳米片.
对钙钛矿纳米晶进行纯化,向上述溶液中加入反溶剂乙酸乙酯,其与甲苯溶液的体积比为3∶1.在8000 r/min 下离心6 min 后倒掉上清液,收集所得沉淀物,并将其分散在1 mL 甲苯溶液中进行进一步表征.
2.3 原位光致发光实验装置及测试
为实时监测纳米晶形成过程中荧光光谱的变化行为,搭建了时间分辨率为100 ms 的原位PL装置.如图1(b)所示,该原位PL 装置包括光谱仪(Maya 2000Pro)、405 nm 激光器、荧光探头、磁力搅拌器、电脑和荧光比色皿.其中光谱仪购于美国的Ocean Insight 公司,405 nm 激光器、荧光探头均购于中国长春新产业光电技术有限公司,荧光比色皿购于谱析光学元件有限公司.
反应在石英比色皿中进行,将其固定在磁力搅拌器后注入甲苯溶液,随后向磁力搅拌的甲苯溶液中注入所需前驱体;同时利用便携式光谱仪的405 nm 激光束(33 mW)定向激发内部形成的发射体,采集纳米晶的发射信号,采集的时间间隔为100 ms.在连续监测20 s 后停止搅拌,转为离线监测,持续监测到2400 s.
3 结果与讨论
3.1 CsPbBr3 PNCs 的相结构和微结构
为验证DDDA 配体的添加比例对晶体结构产生的影响,对合成的具有不同DDDA 添加比例的CsPbBr3PNCs 进行了X 射线衍射(XRD)表征,结果如图2 所示.由XRD 谱图可以看出,未使用DDDA 配体时,CsPbBr3PNCs 样品在10°—50°内有5 个明显的特征衍射峰,分别位于2θ=15.1°,21.2°,30.4°,37.6°,43.6°,对应于单斜相的(100),(110),(200),(211)和(300)衍射面(JCPDS No.18-0364)[10].而添加DDDA 后,所有样品的衍射峰与未添加DDDA 样品的衍射峰相吻合,结果表明DDDA 配体的出现并未改变CsPbBr3PNCs 的相结构.

图2 不同DDDA 比例下所得CsPbBr3 PNCs 的X 射线衍射图Fig.2.X-ray diffraction patterns of CsPbBr3 PNCs synthesized with varying DDDA loadings.
为了探究DDDA 的添加比例对CsPbBr3PNCs尺寸和形貌的调控作用,利用透射电子显微镜(TEM)对所得纳米晶的微结构进行表征,所选样品均为反应时间20 s 时的纳米晶产物,结果如图3所示.图3(a)表明,当反应体系中未使用DDDA配体时,样品的形貌主要呈现为纳米立方块型,其尺寸为(11.51 ± 0.62) nm.从图3(b)—(d)可以清楚地看出,DDDA 配体的出现对形貌有显著影响.当DDDA 的添加比例为25%时,样品中会出现纳米立方块和长条状两种混合形貌,其中以纳米立方块居多,进一步分析发现立方块的尺寸为(9.39 ±0.45) nm,相对于未使用DDDA 的CsPbBr3PNCs样品,其尺寸轻微下降.随着DDDA 添加比例的进一步增加,样品中纳米立方块的数量逐渐变少,而长条状样品的数量逐渐提升.如图3(c)所示,其中长条状样品的宽度和长度分别为(3.06 ± 0.59) nm和(14.49 ± 2.53) nm,而立方块的尺寸进一步减小到(7.40 ± 0.67) nm.对比发现,立方块的尺寸比长条状样品的长度小了近一半,说明体系中存在纳米立方块和纳米片两种形貌,而不是纳米片的顶面和侧面形貌[15].当DDDA 的添加比例为75%时(见图3(d)),纳米晶的形貌主要为细条状,其宽度几乎保持不变,仍然为(3.03 ± 0.53) nm,而长度增大到(21.47 ± 2.33) nm.结果表明,随着DD DA 添加比例的逐渐增加,形貌会从立方块转变为片层结构,同时立方块的尺寸有显著下降.此外通过高分辨透射电子显微镜(HRTEM)还发现,两种形貌的纳米晶都为单晶结构,且结晶质量高(详见补充材料图S1 (online)).据Bekenstein 等[17]报道,具有片层结构的纳米片在干燥过程中极易发生自组装行为,在电子显微镜图中往往处于竖立状态,对于CsPbBr3纳米片来说,其每个原子层厚度约为0.6 nm,因此当DDDA 的添加比例分别为50%和75%时,纳米片的层厚为3 nm,对应层数为5 层.

图3 不同DDDA 比例下所得CsPbBr3 PNCs 的表面形貌表征结果和晶粒尺寸统计结果 (a) 0%;(b) 25%;(c) 50%;(d) 75%Fig.3.TEM images and the corresponding histograms of CsPbBr3 PNCs synthesized with varying DDDA loadings:(a) 0%;(b) 25%;(c) 50%;(d) 75%.
3.2 CsPbBr3 PNCs 的光学性质
结合紫外-可见分光光度计(UV-vis)和光致发光谱仪(PL)进一步探究了DDDA 的添加比例对CsPbBr3PNCs 吸收光谱和荧光光谱(PL 光谱)的影响,结果如图4 所示.所测样品均为20 s 时溶液状态的纳米晶,在未使用DDDA 配体时,吸收光谱图中出现CsPbBr3PNCs 典型的特征吸收信号,且PL 光谱图中观察到尖锐的荧光发射峰,峰位位于509 nm 处,半高全宽(FWHM)为23 nm.当体系中出现DDDA,且随着其添加比例逐渐增加时,出现明显的双荧光峰现象.当DDDA 的添加比例为25%时,其峰位分别位于477 nm 和508 nm 处;当添加比例提高至50%时,其峰位分别 为477 nm 和506 nm.结合TEM 表征和文献[22−24]报道,确定位于蓝光区的荧光峰主要源于纳米片,而位于绿光区的荧光峰主要源于纳米立方块.此外,纳米立方块的峰位会随着DDDA 添加比例的增加,出现轻微蓝移现象,说明立方块的尺寸有轻微下降,与TEM 的结果相一致.当添加比例为75%时,只有单一的荧光峰出现,其峰位在478 nm 处,且从吸收光谱中可以观察到明显的激子吸收峰蓝移现象,结合TEM 表征说明在此条件下主要生成了5 层厚度的纳米片.而在365 nm紫外光照射下,样品的荧光颜色从绿色转变为蓝色(如图4 的插图所示),其变化规律与荧光峰的结果相一致.出现形貌转变很可能是因为DDDA 配体的出现导致反应速率的降低,从而减缓了整个生长动力学过程,因而有利于各向异性纳米晶的生成[25].在合成纳米晶时,观察到未加DDDA 时溶液颜色从无到绿所需时间较短;而出现DDDA 时,溶液早期颜色呈现蓝色,随着添加比例的增加早期出现蓝色的时间更长,而后期溶液颜色会随着反应时间的延长缓慢变绿.
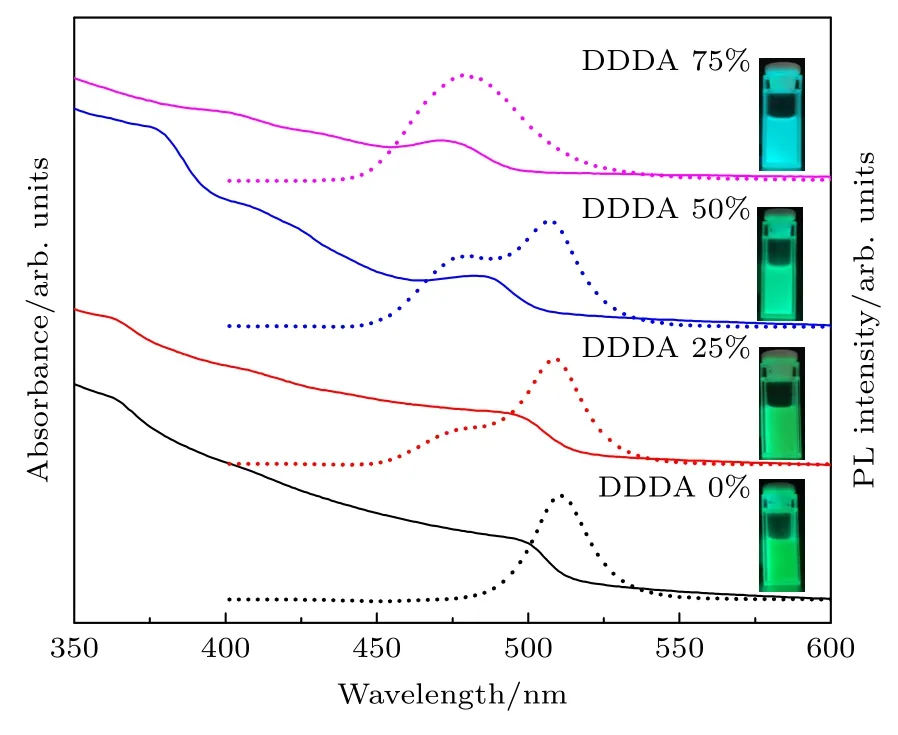
图4 不同DDDA 比例下所得CsPbBr3 PNCs 的吸 收光谱(实线)和荧光光谱(虚线)(插图为365 nm 紫外光照射下的纳米晶溶液照片)Fig.4.Absorption spectra (solid curves) and fluorescence spectra (dotted curves) of CsPbBr3 PNCs synthesized with varying DDDA loadings (Inserts are the photographs of PNCs solutions under 365 nm ultraviolet light illumination).
3.3 CsPbBr3 PNCs 的形成动力学
为深入理解DDDA 配体对CsPbBr3PNCs的形貌调控机理,采用图1(b)中的原位PL 测试平台实时跟踪了CsPbBr3PNCs 快速形成过程中PL 光谱的演变过程.PL 光谱的整个监测时间设置为2400 s,其中前20 s 采取在线监测模式,而21—2400 s 的数据采取离线模式跟踪监测,每个样品点的时间间隔为0.1 s.图5 给出了20 s 内不同DDDA 添加比例下CsPbBr3PNCs 的原位PL 光谱和21—2400 s 的离线PL 光谱(插图).从图5(a)可以清楚地看出,对于没有添加DDDA 配体的样品在0.1 s 时,出现峰位为488 nm 且峰形对称的发射峰,该峰的荧光强度较弱;随着反应的进行(0.1—20.0 s 之间),荧光峰峰位明显红移到509 nm,同时强度显著上升,而PL 峰峰形由不对称变为对称;随着反应时间的进一步增加(t>20 s),峰位和FWHM 保持不变,而峰强继续增加直至不变.

图5 不同DDDA 比例下CsPbBr3 PNCs 在20 s 内的原位PL 图(插图为其在21—2400 s 间的离线PL 图)(a) 0%;(b) 25%;(c) 50%;(d) 75%Fig.5.In-situ PL measurements of CsPbBr3 PNCs synthesized within 20 s under different DDDA loadings,where the inset is the offline PL image during 21–2400 s:(a) 0%;(b) 25%;(c) 50%;(d) 75%.
从图5(b)和图5(c)可知:当体系中分别加入25%和50%的DDDA 配体时,反应液的起始峰位均出现在460 nm 左右,与未添加DDDA 时的样品相比,起始峰位蓝移了28 nm;随着反应的进行,两个体系均出现双荧光峰现象,两个峰的峰位分别在474 nm 和502 nm 左右出现;当反应时间进一步增加,右侧峰会逐渐红移到508 nm 后保持不变,而峰强有显著增加;左侧的PL 峰其峰强逐渐变弱,大约在1800 s 时左侧峰完全消失.为了更好地观察这一双峰现象,本文对实验数据进行了拆分(详见补充材料图S2 和图S3 (online)).当DDDA 添加比例增加至75%时,PL 峰位变化与未添加DDDA 配体时纳米立方块的较为相似,即在整个形成过程中只呈现出单调红移和峰强逐渐上升的现象.不同的是,在该体系反应液的起始峰位在460 nm 左右出现,与DDDA 配体为25%和50%时的一致,说明DDDA 配体的出现对纳米晶的形核阶段和长大阶段都有显著影响.
为了深入揭示纳米立方块与纳米片的形成动力学过程,对图5 的在线PL 谱图进行了系统分析,分别获得了CsPbBr3PNCs 的峰位、FWHM 和峰强随反应时间的变化关系图,如图6 所示.对于未使用DDDA 配体的CsPbBr3PNCs,其整个形成过程根据图6(a)—(c)的峰位、FWHM 和峰强的变化趋势划分为3 个阶段.在第1 阶段(t<2 s),纳米晶的荧光峰峰位在488 nm 处几乎保持不变,峰强稍有增加,而FWHM 呈现出明显的增加趋势,这些现象说明在此阶段纳米晶尺寸非常小,且尺寸变化不大,可以表明该阶段主要发生了前驱体的形核过程.通常晶核尺寸较小、数量少,会导致荧光强度较弱,随着晶核数量的增加其FWHM 值会有所增加,同时峰强会轻微上升.在第2 阶段(2 s≤t≤20 s),随着反应的进行,荧光峰从488 nm 逐渐红移到508 nm 后保持不变,而FWHM 从34 nm减小到23 nm,同时峰强有显著上升,这些现象表明晶核经历了快速长大的过程,且在该过程中尺寸分布逐渐下降,最终获得了尺寸较均一的CsPbBr3纳米立方块.在晶核长大的过程中荧光峰从不对称变为对称,且FWHM 值快速下降到23 nm,该实验现象与Peng 等[26]和Qu 等[27]报道的CdSe 量子点形成过程中PL 的变化规律较为相似,说明晶核的长大经历了尺寸分布聚焦生长机理.当延长反应时间(21 s≤t≤2400 s),纳米立方块的峰位和FWHM 几乎保持不变,只有峰强继续增加.说明LARP 法制备出的CsPbBr3纳米立方块较为稳定,没有发生明显的Ostwald 熟化现象,与Koolyk[28]等报道的高温热注射法合成的CsPbBr3PNCs 结果略微不同,他们利用HRTEM 观察到纳米晶在1—40 s 之间经历了非聚焦生长过程,最终产物的尺寸分布较宽,导致这种生长过程的主要原因是CsPbBr3单体的过快消耗.此外,本文制备的纳米立方块在陈化过程中峰强不断上升,说明陈化过程促进了表面缺陷的有效钝化.

图6 不同DDDA 比例下CsPbBr3 PNCs 的PL 峰位、FWHM 和峰强随反应时间的变化 (a),(d),(g),(j) 峰位;(b),(e),(h),(k) FWHM;(c),(f),(i),(l) 峰强Fig.6.Changes in PL peak position,FWHM,peak intensity of CsPbBr3 PNCs synthesized with varying DDDA loadings as a function of reaction time:(a),(d),(g),(j) Peak position;(b),(e),(h),(k) FWHM;(c),(f),(i),(l) peak intensity.
当反应体系中分别出现25%,50%和75%的DDDA 配体时,如图6(d)—(l)所示,在反应初始阶段(t=0.1 s),所有反应液的荧光峰峰位均在460 nm 左右出现,与未添加DDDA 的样品相比,蓝移了约28 nm.结合TEM 实验结果,在CsPbBr3PNCs 的形成早期-成核阶段,形成了片状晶核,其厚度约为3 层[22].Burlakov 等[29]报道了在成核阶段会与单体竞争成核,当配体与金属离子的键合作用增加时,配体会优先覆盖在较宽的晶体表面,单体只能沿着较窄的面生长,因而实现了各向异性纳米晶的生长.与未使用DDDA 配体的样品相比,本反应体系中额外引入了少量的DDDA 配体,该配体具有双齿羧酸官能团,增加了与Pb2+的相互作用,并降低了体系的反应速率,因而导致成核阶段主要为纳米片结构.随着反应的进行,对于DDDA 添加比例为25%和50%的样品,当PL 峰红移到480 nm 附近时,突然在490—500 nm 之间出现第二峰.为了明确该峰的来源,利用TEM 对DDDA 添加比例为50%的纳米晶样品在刚出现双峰时的形貌进行了表征,结果如补充材料图S4(online)所示.可以观察到片厚为3 nm 的纳米片和边长为4 nm 的纳米立方块两种形貌.说明在反应时间为10 s 左右出现的第二峰为通过均相成核生成的纳米立方块,随着反应时间的增加,会逐渐红移到509 nm,FWHM 值逐渐减小到21 nm,同时峰强呈现出逐渐上升的趋势.当反应时间为20 s 时,第二峰的峰位和FWHM 保持不变,峰强会继续增加,而480 nm 附近源于纳米片的PL 峰峰位几乎保持不变而峰强逐渐减弱.Ng 等[30]同样报道了CsPbBr3纳米片和CsPbBr3纳米立方块两种形貌共存,他们发现随着反应的进行,PL 光谱观察到3 层厚度的纳米片逐渐消失而2 层厚的纳米片逐渐出现,说明纳米片主要沿着厚度方向被逐渐溶解成单体,然后生长到纳米立方块上.不同的是,我们只观察到纳米片逐渐消失的峰.另外,Peng等[31]通过TEM 表征发现,在反应时间为20 s 时纳米片的长度约为14 nm,远远大于最终形成的立方块的尺寸,这些结果表明纳米片会沿着长度方向逐渐溶解后生长到各向同性的纳米立方块上,最终获得的稳定立方块尺寸为9 nm 左右.
从图6 整体来看,与DDDA 为25%和50%的样品相比,当DDDA 的添加比例增加至75%时,最大的区别是整个反应过程中没有出现双峰现象.如图6(j)—(l)所示,在反应早期(t≤1 s),PL 峰峰位基本保持不变,FWHM 下降,峰强缓慢增加,此阶段为纳米片的成核期,成核过程中片的厚度较均一.当反应时间t=1—6 s 时,PL 峰从460 nm 逐渐移动到478 nm 左右,而峰强快速上升的同时FWHM 也增加,说明纳米片长大变厚过程中,其厚度均一性变差,也可能是出现少量的纳米立方块所致,由于数量较少没有出现独立的纳米立方块所属的荧光峰.此外,还可以从图6(j)—(l)明显看出,纳米片在5—20 s 间处于稳定期,其峰位、FWHM 和峰强均保持不变.当反应时间进一步增加,纳米片会从各向异性逐渐生长为各向同性的纳米立方块,其最终峰位稳定在505 nm (详见补充材料图S5 (online)).结果表明,当DDDA 的添加量进一步增加时,由于配体在较宽晶面的覆盖及生长速率的进一步降低,单体主要沿着纳米片的长度方向生长,而厚度方向保持不变,最终生成了大量纳米片,且生成的纳米片在20 s 之内较为稳定.但随着反应时间的延长,通过纳米片长度方向上破碎溶解下来的单体会继续生长到厚度方向,最终生成各向同性的稳定纳米立方块,其最终尺寸为7—8 nm[31](详见补充材料图S6 (online)).实验结果表明,在CsPbBr3PNCs 的成核和生长早期,通过调节DDDA 的添加比例获得了纳米片,抑制了纳米立方块的形成,该过程由动力学控制;而随着反应时间的延长,在生长后期发生了纳米片结构向纳米立方块的转变,该过程由热力学控制.可见,在CsPbBr3PNCs 的形成过程中,热力学和动力学共同控制着纳米晶的形貌、尺寸和均一性.
为了清楚地描述CsPbBr3PNCs 的形成动力学过程,图7 给出了反应时间为0—2400 s 的纳米晶形成机理示意图.DDDA 的出现对纳米晶的形成过程有显著影响.

图7 CsPbBr3 钙钛矿纳米晶的生长机理示意图Fig.7.Schematic presentation of growth mechanisms of CsPbBr3 PNCs.
1)当DDDA 的添加比例为0%时,CsPbBr3纳米立方块在20 s 内形成,纳米立方块经历了快速成核和尺寸分布聚焦生长两个过程,最终立方块的边长为11 nm 左右;在陈化阶段,纳米立方块没有发生Ostwald 熟化现象,尺寸和尺寸均一性保持不变,而发光强度有显著提升.
2)当反应体系中出现25%和50%的DDDA配体时,纳米晶的整个形成过程变缓,这是因为DDDA 配体具有双齿羧酸官能团,其与Pb2+发生较强的配位作用,使得纳米晶垂直方向的生长速率降低,而水平方向的生长速率增加,因而在成核阶段出现了片层结构的晶核.随着反应的进行,体系中会出现通过均相成核生成的纳米立方块,且与纳米片同时存在.而纳米片具有不对称结构,会沿着长度方向逐渐分解后再结晶到纳米立方块上,最终产物为尺寸均一的纳米立方块.
3)当DDDA 配体的添加比例增加至75%时,由于反应速率的进一步降低和DDDA 与Pb2+存在的较强相互作用,因而在成核和生长早期仍然以片层结构为主,片的厚度约为3 nm (约为5 层),说明在形成早期完全抑制了纳米立方体的形成.随着反应时间的延长,纳米片会通过减小长度和逐渐增加厚度(即溶解-再结晶)的方式转变为尺寸较为均一的纳米立方块.
综上所述,通过DDDA 配体的添加降低了反应体系的生长速率,从纳米晶的形成动力学调控获得了纳米片,而纳米片通过热力学控制逐渐转变为尺寸和尺寸均一性几乎一致的纳米立方块.
4 结论
采用配体辅助再沉淀法制备了CsPbBr3钙钛矿纳米晶,考察了DDDA 配体的添加比例对纳米晶的相结构、微结构和光学性质的影响.结果表明,DDDA 的出现,对纳米晶的相结构没有影响;随着DDDA 添加比例的增加,形貌逐渐从纳米立方块转变为纳米片,同时激子吸收峰和荧光峰出现明显蓝移现象.结合搭建的具有超高时间分辨能力的原位PL 装置,揭示了纳米晶中没有DDDA 时只经历快速成核和尺寸分布聚焦生长,反应结束时获得的纳米立方块的尺寸约为11 nm;当添加比例分别为25%和50%时,前驱体快速形成片层晶核,核生长过程中出现纳米立方块和纳米片混合形貌,纳米片在较长时间后会发生溶解-再结晶转变为各向同性的纳米立方块;当DDDA 添加比例为75%时,在5 s 内经历了快速形成片层晶核和晶核缓慢生长为5 层厚度的纳米片,稳定了15 s 后逐渐转变为尺寸较为均一的纳米立方块.通过调节DDDA的添加比例而降低了纳米晶体的生长速率,从而实现了动力学控制获得不同形貌的钙钛矿纳米晶.
