负载型Pd-Cu催化剂的制备及富氢气氛下CO优先氧化性能
2022-05-25赵婉君王永钊赵永祥
赵婉君,李 潇,党 慧,王永钊,赵永祥
(1.山西大学精细化学品教育部工程研究中心,2.化学化工学院,太原030006)
氢气作为清洁能源在化工领域具有巨大的应用潜力,但是通过甲烷重整和水煤气变换(WGS)反应产生的H2中通常含有少量的CO(体积分数约1%).在质子交换膜燃料电池(PEMFCs)、合成氨和加氢精制等化工过程中,微量CO会使Pt电极或催化剂中毒失活,因此进一步消除微量CO至关重要[1].富氢气氛中CO优先氧化(CO-PROX)是一种极具吸引力的H2净化方法,该方法可显著降低CO消除过程中的能量损失,已被公认为消除富氢体系中微量CO的优选方案[2].
CO优先氧化催化剂体系包括贵金属催化剂和非贵金属氧化物催化剂.贵金属催化剂中铂族催化剂(Pt[3],Pd[4],Ru[5]和Rh[6]等)和Au[7~9]基催化剂因其良好的CO,O2吸附能力和催化性能备受关注.铂族催化剂虽然具有较高的反应活性和稳定性,但存在价格昂贵、反应温度较高和选择性相对较低等问题.Au基催化剂在低温(<100℃)下具有较高的CO转化率,但由于在反应条件下易形成碳酸盐物种或在水汽存在情况下极易失活,使其应用受到一定限制[10].非贵金属氧化物催化剂中CuO-CeO2在CO优先氧化反应中表现出良好的催化性能,但其抗H2O,CO2毒化能力稍差[11~13].相对于上述催化剂体系而言,Pd基催化剂表现出更低的催化活性和选择性.Zlotea等[14]利用同步辐射X射线吸收光谱(XAS)探究了Pd/SiO2-Al2O3在CO氧化和CO优先氧化反应中的催化活性,发现该催化剂具有极低的CO优先氧化反应活性,即使在250℃时CO最高转化率也仅有15%,这是由于活性Pd物种在低温(<300℃)下易形成PdHx,不利于CO的吸附活化.Boukha等[15]发现Pd/羟基磷灰石的CO-PROX催化性能较差,这同样是由于H2比CO更容易在金属Pd上吸附和氧化.Pozdnyakova等[16]利用原位技术研究了Pd/CeO2的CO-PROX催化性能,发现在低温下形成的PdHx会抑制CO氧化.尽管PdHx在高温下分解可使CO转化率适当增加,但同时H2的氧化导致CO2选择性降低.为了提高Pd基催化剂的CO-PROX催化活性与选择性,许多研究者进行了积极而有益的探索.Cao等[17]采用区域选择性原子沉积的方法合成了核壳结构的Pd@Pt纳米颗粒,发现与单一金属纳米颗粒相比,Pd@Pt具有更好的CO-PROX催化活性与选择性,这可归因于Pt包覆的Pd催化剂上CO氧化的能垒显著降低.Zhang等[18]通过软模板法合成了具有中空结构和多孔壁的Pd-Pt合金纳米笼,并用于CO-PROX反应,发现适当尺寸的Pd-Pt纳米笼有利于提高CO转化率,同时纳米笼特殊的中空结构可抑制H2氧化以提高CO2选择性.Miguel-García等[19]利用溶剂热还原法制得聚乙烯吡咯烷酮(PVP)保护的Pd纳米颗粒,然后通过浸渍法得到Pd-Al2O3催化剂并用于CO优先氧化反应,结果表明,在H2O和CO2存在时,CO在180℃时可实现完全转化且具有较高的选择性.分析认为,由于PVP长链上羰基产生的电荷转移络合物削弱了Pd纳米颗粒周围的电子云密度,有利于CO吸附活化,同时抑制了H2的竞争吸附与氧化.Morfin等[20]将Ir-Pd纳米合金负载在非晶态二氧化硅-氧化铝上并用于催化CO-PROX,发现合金中Ir和Pd之间的协同作用可抑制PdHx的形成,提高了催化剂的催化活性和选择性.虽然上述策略在一定程度上削弱了H2的竞争吸附,但由于反应温度较高仍不足以完全避免H2氧化反应,CO2选择性一般低于50%.目前Pd基催化剂仍存在低温下催化活性差和高温下CO2选择性低的问题,因此,探索新型的CO优先氧化Pd基催化剂体系具有重要意义.
负载型Wacker催化剂(Pd-Cu/载体)长期以来被广泛研究,并用于含水条件下低温CO氧化反应[21~24],但其富氢条件下CO优先氧化反应性能鲜见报道.载体材料对负载型Wacker催化剂织构、活性相结构、组成及其催化性能具有重要影响.其中两性氧化物Al2O3因其同时具有酸碱性[25]、高比表面积以及与活性组分之间的强相互作用[26],常作为负载型催化剂载体[24,27].无定形SiO2被广泛用作非均相催化剂的载体,通过修饰其表面硅羟基可使活性位点处于高分散状态,进而提高催化性能[28].而C3N4由于其特殊的结构而具有较高的热稳定性、可调节的电子结构和氧惰性,且易于制备[29],目前已在光催化、燃料电池、储氢和环境保护等诸多领域受到广泛研究.相比较而言,3种载体的表面性质和结构各不相同,其中Al2O3和SiO2载体中均含有表面羟基,而C3N4特殊的结构中则不存在羟基;同时,Al2O3比SiO2表现出更强的表面碱性.因此为了探究载体对负载型Pd-Cu催化剂CO优先氧化反应性能的影响,本文分别选用Al2O3,SiO2和C3N4为载体,制备了负载型Pd-Cu催化剂,并考察其富氢气氛中CO优先氧化反应性能,同时通过多种表征技术探讨了载体性质与其诱导的催化剂结构性能变化的关系,以期为新型高效CO-PROX催化剂的设计提供参考.
1 实验部分
1.1 试 剂
PdCl2‧2H2O、正硅酸乙酯(TEOS)和冰醋酸,购于国药集团化学试剂有限公司;CuCl2‧2H2O和NH3‧H2O,购于天津市天力化学试剂有限公司;Al(NO3)3‧9H2O和C2H5OH,购于天津市大茂化学试剂厂;双氰胺,购于阿拉丁科技(中国)有限公司.上述试剂均为分析纯.
1.2 载体的制备
采用溶胶-凝胶法制备Al2O3载体.将一定量Al(NO3)3‧9H2O溶于150 mL去离子水中,并置于70℃的水浴中,缓慢滴加NH3‧H2O,边滴加边剧烈搅拌直至体系pH约为9,形成半流动状态的白色溶胶,然后老化1 h,于70℃干燥24 h,在500℃焙烧1 h,最终得到Al2O3载体.
采用溶胶-凝胶法制备SiO2载体.将25 mL TEOS溶于50 mL乙醇中,加入16.6 mL冰醋酸和24 mL去离子水,在60℃下充分搅拌,室温老化24 h,形成凝胶,再经65℃干燥得到近似透明的SiO2原粉,将原粉置于马弗炉中于300℃焙烧2 h后得到SiO2载体.
采用热聚合法制备C3N4载体.将一定量双氰胺置于洁净坩埚中,在空气气氛下置于马弗炉中于550℃焙烧3 h,随后将得到的样品自然冷却,研磨后即为C3N4载体.
1.3 催化剂的制备
采用浸渍法制备负载型Pd-Cu催化剂.将1.82 g CuCl2‧2H2O溶解在2 mL 9.97 mg/mL PdCl2溶液中,向其中加入5 g Al2O3(为保证等体积浸渍,对于SiO2而言,在浸渍过程中额外加入了1.5 mL蒸馏水,而C3N4上则分3次浸渍),老化3 h,于120℃干燥6 h,然后在空气气氛下于300℃焙烧2 h,最终得到3种不同的催化剂,分别记为PC-Al2O3,PC-SiO2和PC-C3N4.
1.4 催化剂的表征
X射线衍射(XRD)表征在室温下Bruker D8-Advance型X射线粉末衍射仪(瑞士布鲁克公司)上进行,采用CuKα射线(λ=0.15418 nm),Ni滤波,5°~80°扫描,扫描速率2.4°/min,万特探测器检测.采用Bruker Tensor 27型傅里叶变换红外光谱仪(FTIR,瑞士布鲁克公司)对催化剂进行红外光谱测定,样品与溴化钾(质量比为1∶100)混合,分辨率4 cm−1,400~4000 cm−1扫描.低温N2物理吸附测定在Micromeritics ASAP-2020型物理吸附仪(美国Micromeritics公司)上进行.催化剂预先在150℃真空条件下脱气预处理5 h,然后在液氮下进行氮气的吸附-脱附测定,分别用BET公式和BJH模型计算样品的比表面积和孔径分布.氢气程序升温还原(H2-TPR)测试样品用量为30 mg,混合气组成为5%H2-95%氮气(体积分数),以10℃/min升温速率程序升温至600℃,通过CuO定量还原为单质铜来校准[30].二氧化碳程序升温脱附(CO2-TPD)测试样品用量为100 mg,在He气气氛中升温至300℃并恒温1 h,再冷却至50℃后吸附CO2脉冲气体直至饱和,最后升温至600℃.在相同测试条件下扣除背景后得到CO2-TPD曲线[31].催化剂的X射线光电子能谱(XPS)在ESCALAB 250型光谱仪(美国Thermo Fisher Scientific公司)上测定,使用AlKα辐射作为激发源.以碳的C1s结合能作为基准进行校正.原位漫反射傅里叶变换红外光谱(in situDRIFTS)在Nicolet is50型傅里叶变换红外光谱仪(美国Thermo Fisher Scientific公司)上进行测试,样品用量为45 mg,样品压片后放入反应池中,在300℃下用氮气吹扫1 h,随后降温至30℃,此时扫描红外光谱作为背景;最后将氮气切换为原料气(1%CO,1%O2,3.3%H2O,50%H2,氮气为平衡气)保持60 min,并间隔记录样品的红外谱图.
1.5 催化活性的评价
CO优先氧化活性评价在连续流动微反应装置上进行.催化剂用量为0.3 g,原料气组成(体积分数)为1%CO,1%O2,3.3%H2O,50%H2,氮气为平衡气,空速(GHSV)为6000 h−1.反应前未对催化剂进行任何预处理,反应温度为30℃.采用GC-930型气相色谱仪(上海海欣色谱仪器有限公司)分析原料气及产物组成,O2经5A分子筛柱分离后由热导检测器(TCD)检测,CO和CO2经TDX-01柱分离后通过甲烷转化炉转化成甲烷,再借助火焰离子化检测器(FID)检测.催化剂的CO转化率(X,%)和CO2选择性(S,%)根据以下公式计算:

式中:COin和O2in为反应前混合气中CO的含量(体积分数,%)和O2的含量(体积分数,%),COout和O2out为反应后混合气中CO的含量(体积分数,%)和O2的含量(体积分数,%).
2 结果与讨论
2.1 催化活性
图1为富氢气氛下不同Pd-Cu催化剂上CO优先氧化活性的评价结果.可以看出,3种催化剂的CO转化率明显不同:PC-Al2O3初始CO转化率高达57%,当反应进行30 min后略有下降,然后在所考察的反应时间范围内可维持在约27%,这一结果优于单Pd催化剂上的CO-PROX催化性能(Zlotea等[14]报道的Pd/SiO2-Al2O3在250℃时CO最大转化率仅为15%).PC-SiO2上CO初始转化率(52%)与PC-Al2O3接近,但随时间延长转化率急剧下降,在反应45 min后几乎无活性.PC-C3N4则始终表现出极低的CO转化率.值得一提的是,当反应温度为30℃时,3种催化剂上CO2选择性均为100%.可见,虽然富氢气氛下3种负载型Pd-Cu催化剂上CO优先氧化催化活性不同,但低温下该类型催化剂可抑制H2的竞争性氧化反应.
2.2 XRD表征
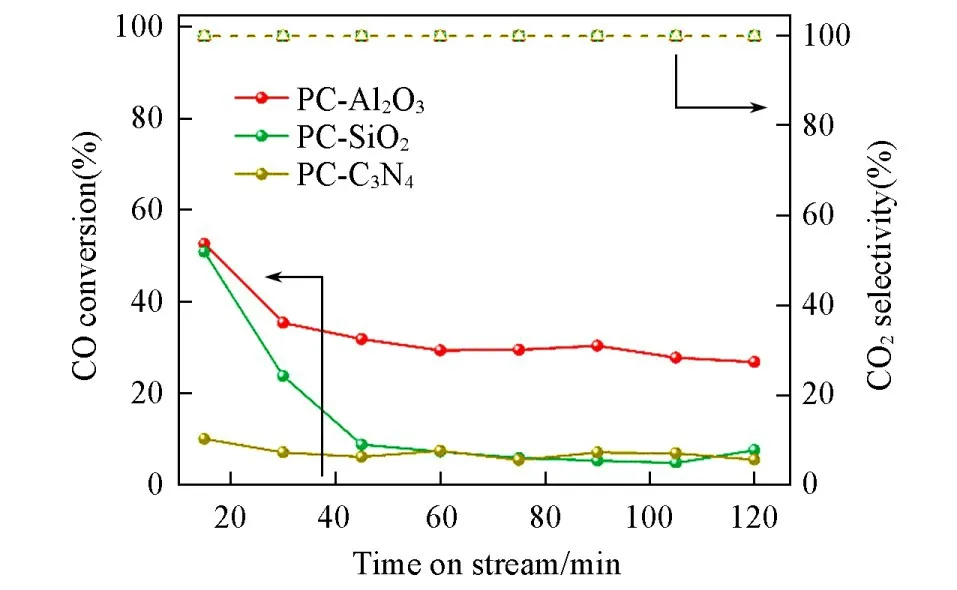
Fig.1 Catalytic performance of catalysts for preferential CO oxidation in rich H2 stream
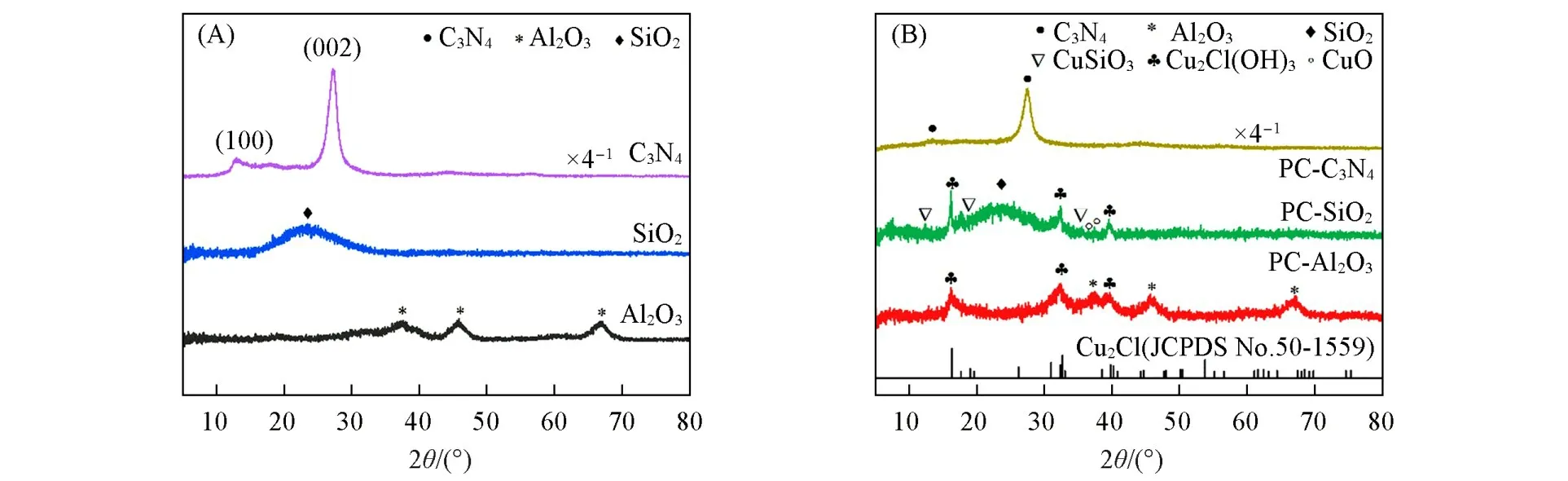
Fig.2 XRD patterns of supports(A)and catalysts(B)
图2(A)为不同载体的XRD谱图,其中Al2O3在2θ=37.6°,45.9°和67.3°处的衍射峰对应于γ-Al2O3的标准卡片(JCPDS No.51-0769);SiO2在2θ=22.5°处出现的弥散衍射峰与无定形SiO2的标准卡片(JCPDS No.44-0696)相符;C3N4在2θ=12.8°和27.3°处存在2个衍射峰,表明所得样品为石墨相氮化碳,这与g-C3N4的标准卡片(JCPDS No.50-1512)一致.图2(B)为催化剂的XRD谱图,PC-Al2O3和PC-SiO2均在2θ=16.1°,32.3°和39.5°处出现衍射峰,这与Cu2Cl(OH)3的标准卡片(JCPDS No.50-1559)相符,表明这两种催化剂中均存在Cu2Cl(OH)3相.与PC-SiO2相比,PC-Al2O3中该物相衍射峰峰形弥散且强度相对较低,表明Al2O3上Cu2Cl(OH)3具有更好的分散性.此外,二者还存在少量的CuO(JCPDS No.45-0937)物种.不同的是,PC-SiO2在2θ=12.2°,18.2°,35.5°处还出现了微弱的衍射峰,对应CuSiO3(JCPDS No.32-0346).PC-C3N4催化剂中未观察到铜物种的特征衍射峰,与载体相比,PC-C3N4中载体的衍射峰强度明显减弱,这可归因于Cu物种嵌入C3N4基质中形成了与C3N4直接配位的基体[32].3种催化剂中均未检测到Pd物种的特征衍射峰,表明Pd物种高度分散于催化剂中.
2.3 氮气物理吸附-脱附表征
图3(A)和(B)分别为不同载体和Pd-Cu催化剂的氮气物理吸附-脱附曲线.Al2O3的氮气吸附-脱附曲线对应于Ⅳ型等温线[33],在中压区出现H4型回滞环,表明Al2O3中具有一定的介孔结构.SiO2的吸附-脱附等温线趋势与文献[34]中报道相一致,属于Ⅰ型等温线[34],说明SiO2中存在一定量的微孔结构.就C3N4而言,其吸附-脱附曲线在相对压力(p/p0=0.8~0.99)较高处出现H3型回滞环[35],表明C3N4中存在由薄片堆积形成的层状结构.引入活性组分后,3种催化剂的氮气吸附-脱附曲线均呈现出与载体相似的趋势.
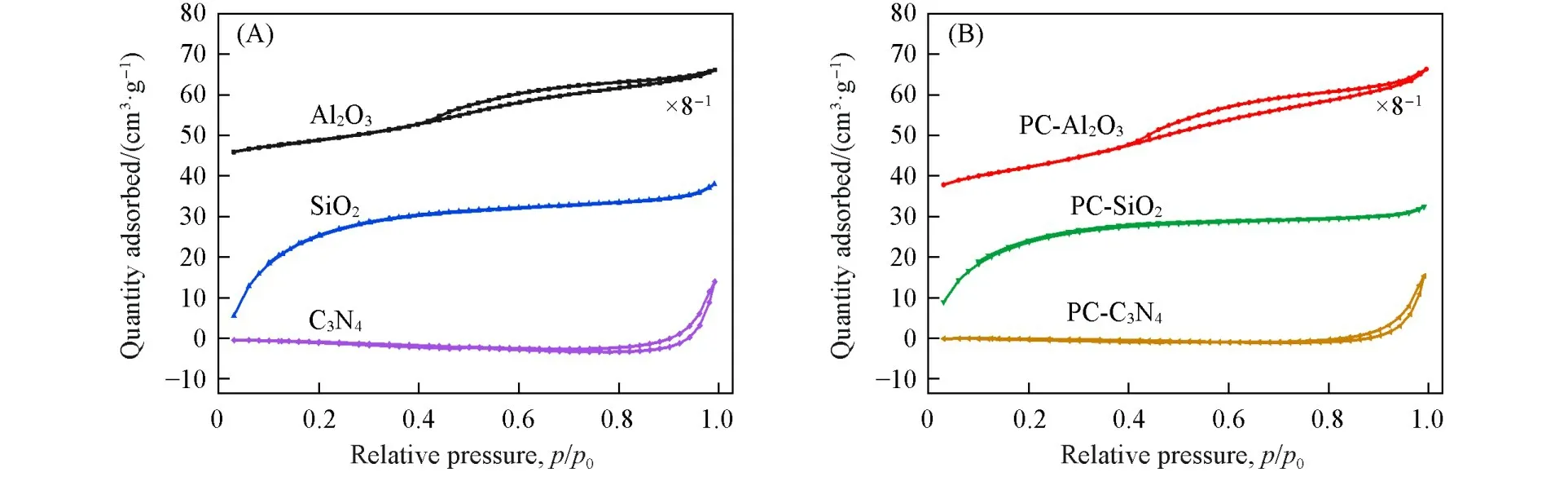
Fig.3 N2 adsorption⁃desorption isotherms of supports(A)and catalysts(B)
表1列出了载体和催化剂的织构数据.可以看出3种载体的比表面积、孔容以及平均孔径存在明显差异:SiO2的比表面积最大,Al2O3次之,C3N4比表面积最小;孔容按照Al2O3>SiO2>C3N4顺序依次减小;而平均孔径则出现相反的规律.与相应载体相比,PC-Al2O3,PC-SiO2的比表面积和孔容均减小,这可归因于部分活性组分进入载体的孔道所致.而PC-C3N4的比表面积、孔容均大于C3N4载体,平均孔径则有所减小.结合3种催化剂的活性评价结果,比表面积较大的PC-Al2O3和PC-SiO2的CO初始转化率优于比表面积最小的PC-C3N4,可以推测负载型Pd-Cu催化剂的织构性质可能是影响其催化活性的原因之一;但对比PC-Al2O3和PC-SiO2,发现前者在长时间反应过程中的催化活性优于后者,而前者的比表面积低于后者,表明催化剂的织构性质并不是影响催化活性的主要因素.
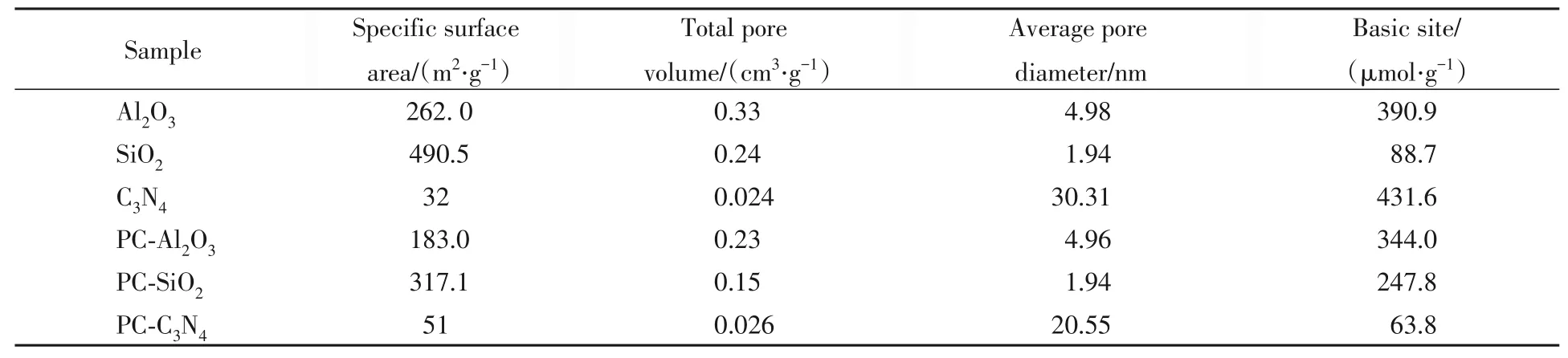
Table 1 Textural properties and basic sites of different supports and catalysts
2.4 红外表征
图4(A)和(B)分别为不同载体和催化剂的FTIR谱图.图4(A)中Al2O3在约3445 cm−1附近的吸收峰对应于物理吸附水和表面羟基的伸缩振动,约1635 cm−1处的吸收峰对应H—O键的弯曲振动[36];SiO2同样在3455和1640 cm−1附近出现了吸收峰,这可归属于硅羟基和表面吸附水的O—H键伸缩和弯曲振动,此外,1076,808和463 cm−1处的峰均为SiO2中Si—O—Si结构的红外吸收峰[37].C3N4的红外谱图在3167 cm−1附近出现的吸收峰对应于N—H键伸缩振动,表明其层状结构边缘存在未完全缩合的氨基基团[38];2155 cm−1附近的峰对应七嗪环中C≡N基团的伸缩振动,1641~1242 cm−1之间较宽的强峰,则是由七嗪环中C—N和C=N杂环的伸缩振动引起的,806 cm−1处出现的尖锐的强峰可归属于3-s-三氮烷环单元的伸缩振动[32].
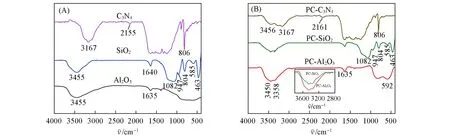
Fig.4 FTIR spectra of supports(A)and catalysts(B)
如图4(B)所示,PC-Al2O3和PC-SiO2的红外谱图在3445 cm−1附近出现载体的羟基红外峰,此外,在3358和3450 cm−1附近还可以观察到额外的红外峰[如图4(B)中两个相邻的小尖峰],结合XRD表征结果,该吸收峰归属于Cu2Cl(OH)3物种中OH基团的不对称和对称伸缩振动[39].PC-C3N4在3456 cm−1处出现的吸收峰可对应—OH的伸缩振动[40],表明负载活性组分后其表面也产生了物理吸附的水分子;与载体C3N4中杂化C≡N的2155 cm−1峰位相比,PC-C3N4中该吸收峰蓝移至2161 cm−1附近,这可归因于C3N4基体与Cu物种之间的强相互作用[32].此外,从图4(B)插图中可见,PC-Al2O3在3358和3450 cm−1处的吸收峰强度明显强于PC-SiO2,表明PC-Al2O3中存在相对更高含量的Cu2Cl(OH)3物种[31].
2.5 H 2-TPR表征
图5为不同催化剂的H2-TPR谱图.大体上可将温度范围分为低温区(α)和高温区(β).PC-Al2O3(208℃,225℃)与PC-SiO2(203℃,260℃)在低温区的还原峰可以分别归属于Pd物种与高分散Cu2Cl(OH)3和微晶态Cu2Cl(OH)3的共还原[41].PC-Al2O3在高温区318℃的还原峰可归属于部分孤立CuO物种的还原[24].PC-SiO2高温还原峰(376℃)则对应体相CuO和CuSiO3的还原[42~44].PC-C3N4的还原峰趋势与文献[32]中报道一致,在图5插图中观察到236℃处出现一个极弱的还原峰,对应于部分表面Cu2+到Cu+的还原;而高于450℃的区域则对应于Cu2+/Cu+还原为Cu0(但不排除该过程中伴随着C3N4的热分解)[32].从还原温度可看出,与PC-Al2O3,PC-SiO2相比,C3N4基质中的Cu物种相对较难还原,这与Cu物种嵌入C3N4基质中形成配位基体有关.表2列出了不同催化剂的H2消耗量.从表2中可以看出,PC-Al2O3的实际耗氢量略小于理论耗氢量(1910μmol/g),大于PC-SiO2的耗氢量,二者均远大于PC-C3N4(由于PC-C3N4在高温处Cu物种的还原伴随着C3N4基质的分解,此处未考虑其高温耗氢量).同时,PC-Al2O3低温区2个还原峰的耗氢量明显高于PC-SiO2且还原温度更低,表明PC-Al2O3催化剂中不仅存在更多数量的Cu2Cl(OH)3,Pd,Cu物种更易还原且具有更强的相互作用,这与相应的XRD和FTIR表征结果一致.总体而言,在3种负载型Pd-Cu催化剂中PC-Al2O3具有相对更强的氧化还原性能.
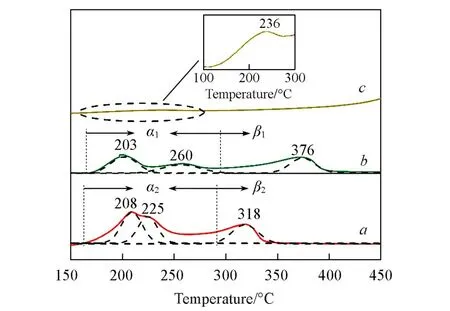
Fig.5 H 2⁃TPR profiles of catalysts PC⁃Al2O3(a),PC⁃SiO2(b)and PC⁃C3N4(c)

Table 2 H 2 consumption of catalysts
2.6 CO2-TPD表征
图6(A)和(B)分别为不同载体和催化剂的CO2-TPD谱图.Al2O3在低温160℃左右出现的CO2脱附峰对应于弱碱性位点[45].负载活性组分后,CO2脱附峰温度略微降低,同时对应的峰面积稍有减小,表明PC-Al2O3催化剂碱性减弱,这可能是由于CuCl2的碱性弱于Al2O3,引入大量Cu物种之后使其碱性减弱[46].SiO2在75~160℃温度范围内出现小的CO2脱附峰,表明SiO2载体中存在相对较弱的碱性位点[45].与载体相比,PC-SiO2的CO2脱附峰向高温方向移动,峰面积增大,表明Pd,Cu的加入使其样品的碱性稍有增强.C3N4载体在较宽温度区间(70~500℃)出现CO2脱附峰,表明C3N4中含有多种形式的碱性位点,且碱性相对于Al2O3和SiO2更强.PC-C3N4表面除弱碱性位点外,在高于400℃出现了明显的脱附峰,表明其表面还存在相对较强的碱性位点.结合FTIR表征结果,其可能是由于C3N4中C—N—C六元环中的≡N、表面的=NH或—NH2基团引起的[29].表1中列出了不同载体和催化剂的碱性位点数量,3种载体的弱碱性位点数量按下列顺序依次减小:C3N4>Al2O3>SiO2.负载Pd,Cu后,PC-Al2O3上弱碱性位点数量减少,而PC-SiO2上增多.C3N4虽具有相对更强的碱性,但由于氮原子的电负性较低,吸电子能力较弱,其更容易给出电子,从而表现出较强的配位能力.因此Cu更易与C3N4中N原子结合形成Cu—N键,不利于形成Cu2(OH)3Cl物种,最终表现为Cu物种嵌入C3N4基质中形成了与C3N4直接配位的基体,导致其碱性明显减弱.结合XRD和FTIR结果可知,碱性较强的Al2O3载体更有利于活性Cu2Cl(OH)3物种的形成.
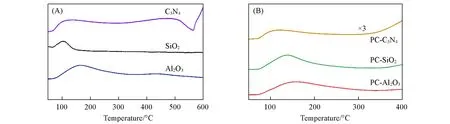
Fig.6 CO 2⁃TPD profiles of supports(A)and catalysts(B)
2.7 XPS表征
图7(A)和(B)分别为3种催化剂的Pd3d和Cu2p3/2的XPS谱图.在图7(A)中,3种催化剂在约337.8和342.8 eV处出现了相似的特征峰,可分别对应于Pd3d5/2和Pd3d3/2,表明Pd物种主要以Pd2+形式存在[22].如图7(B)所示,3种催化剂在约934.5和932.2 eV附近出现的特征峰可分别归属于Cu2+和Cu+物种[41].其中与PC-Al2O3和PC-SiO2相比,而PC-C3N4中Cu2+和Cu+物种的结合能相对较高,这主要与Cu和C3N4基体之间的配位作用有关.进一步对Pd3d和Cu2p的XPS谱图进行分峰拟合,根据峰面积定量分析催化剂表面的物种,不难发现PC-Al2O3和PC-SiO2表面的Cu2+含量大于Cu+,而PC-C3N4中Cu+的含量明显大于Cu2+.3种催化剂表面的Pd,Cu相对原子比用Pd2+/(Cu++Cu2+)来表示,计算结果如表3所示.PC-Al2O3催化剂表面的Pd,Cu相对原子比明显大于PC-SiO2和PC-C3N4,表明PC-Al2O3催化剂表面具有更多的Pd2+物种.同时,计算了催化剂表面Cu物种的相对含量,用Cu+/(Cu++Cu2+)来表示.经比较发现,PC-C3N4催化剂的Cu+/(Cu++Cu2+)原子比远大于其它2种催化剂,这主要是由于Cu-N配位过程中形成大量的Cu+物种;PC-Al2O3中Cu+/(Cu++Cu2+)的原子比(0.212)大于PC-SiO2(0.115),表明PC-Al2O3中含有更多的Cu+物种.
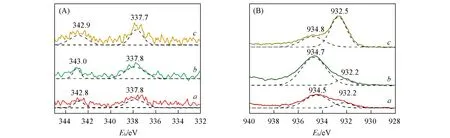
Fig.7 XPS spectra of Pd 3d(A)and Cu2p3/2(B)of catalysts PC⁃Al2O3(a),PC⁃SiO2(b)and PC⁃C3N4(c)
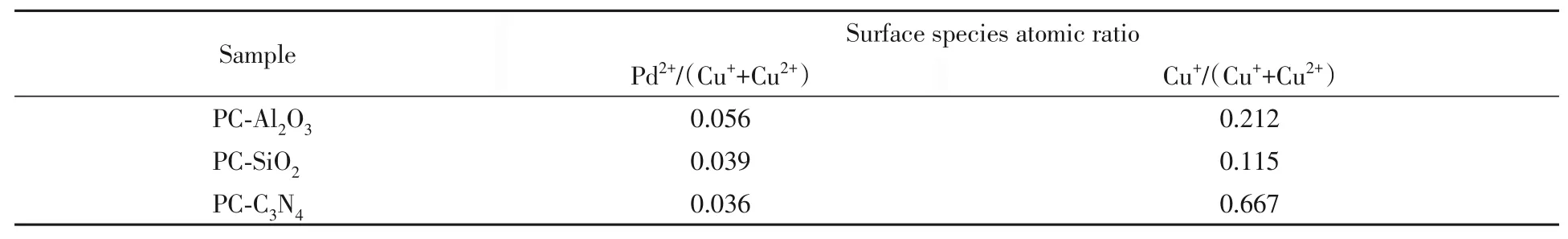
Table 3 Parameters of catalysts obtained from XPS analysis
2.8 In situ DRIFTs表征
图8为原料气在3种催化剂上的In situDRIFTS谱图.如图8(A)所示,2162 cm−1处的峰与CO线性吸附在Pd2+相关(Pd2+-CO),1990~2000和1930~1940 cm−1处的峰可分别对应CO在金属态Pd(Pd2-CO)和Pd+(Pd+-CO)上的桥式吸附[47].在2123和2131 cm−1处的强峰分别对应SⅠ和SⅡ2种吸附中心上的Cu+-CO络合物[24].
从图8(A)可见,在前30 min内随着吸附时间的延长,PC-Al2O3上Pd2+-CO吸收峰消失,Pd+-CO和Pd2-CO吸收峰逐渐增强,表明Pd2+物种易被CO还原.与Pd2+相比较而言,由于Pd+具有更丰富的电子,有利于稳定CO反馈π键而表现出更强的活化CO能力[21],但当Pd+被CO还原为Pd0后,Pd0物种在大量H2存在时极易形成PdHx[15,16],不利于Pd0再氧化,从而抑制了CO的吸附活化,这可能是反应初期CO转化率逐渐下降的原因[16].此外,Cu物种上CO特征吸收峰在吸附过程中Cu+-CO的吸收峰(2131 cm−1)向低波数方向(2123 cm−1)移动,该红移现象意味着Cu+-CO的吸附中心从SⅡ吸附中心逐渐迁移至SⅠ吸附中心,而SⅠ吸附中心上富电子的Cu+物种更易氧化为Cu2+.此时,Cu2+的形成又进一步促进还原态Pd0物种的氧化,使得催化循环继续进行.因此,PC-Al2O3中Pd,Cu物种间具有较强的相互作用,可以在一定程度上促进部分Pd0的再氧化,避免催化活性进一步降低.
从图8(B)可知,随着吸附时间延长,PC-SiO2中Pd2+-CO峰强度逐渐减弱,Pd2-CO吸收峰强度逐渐增强,同时在1950 cm−1处出现额外的吸收峰,对应CO在还原态(Pd*)上的吸附(Pd*-CO)[16].Pd*-CO吸附峰的位置(1950 cm−1)低于Pd+-CO(1930~1940 cm−1),表明Pd*不如Pd+对CO具有更强的活化能力.另外,PC-SiO2中Cu+-CO的吸附峰对应SⅡ吸附中心,其峰强度远低于PC-Al2O3中Cu+上CO吸附峰[见图8(D)],同时在Pd2+被还原过程中该峰位置未发生偏移,表明PC-SiO2上Pd,Cu物种间的相互作用较弱,且Cu+物种数量远低于PC-Al2O3,这与TPR,XPS等表征结果一致.可见,PC-SiO2中Cu2+不利于Pd物种再氧化而导致Cu+含量随之减少.
从图8(C)中可以看出,PC-C3N4中Pd2+-CO的吸收峰强度较弱,表明C3N4上的Pd2+不利于CO吸附活化.同时与PC-Al2O3和PC-SiO2相比,PC-C3N4中Cu+-CO的吸收峰极弱,可见PC-C3N4中入C3N4基质的Cu物种同样不利于CO的吸附.因此,PC-C3N4表现出极低的催化活性.
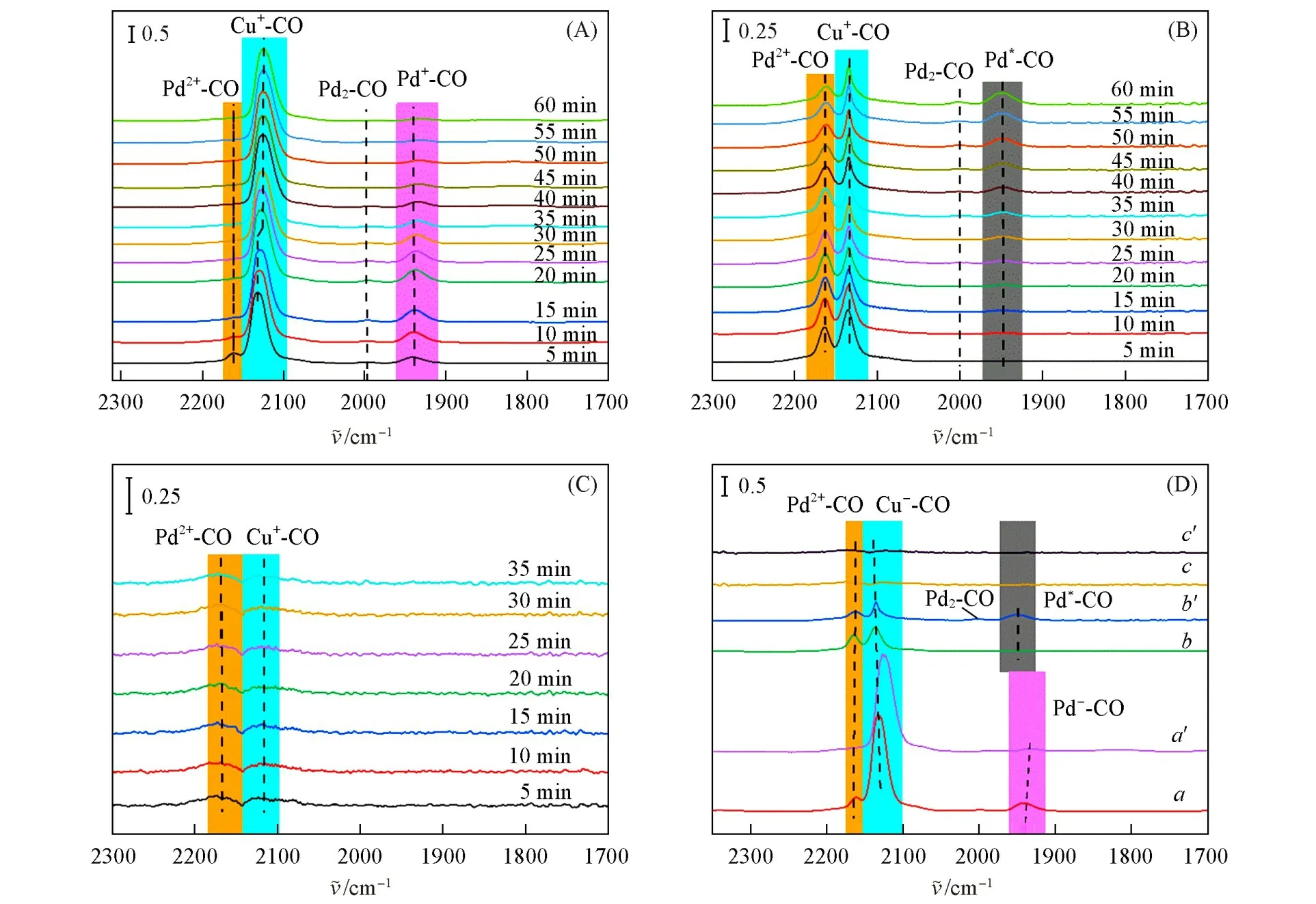
Fig.8 In situ DRIFTs spectra of PC⁃Al2O3(A),PC⁃SiO2(B),PC⁃C3N4(C),three catalysts after 5 min(a,b,c),60 min(a′,b′)and 35 min(c′)(D)under 1%CO⁃1%O2⁃48%H 2⁃N2 at 30℃
结合上述表征结果可知,Al2O3和SiO2载体负载Pd,Cu后,均可形成Cu2Cl(OH)3活性相,而C3N4载体上则未出现该物种.究其原因,与载体的表面羟基性质和结构有关.众所周知,活性氧化铝[图9(A)]在有水环境下会发生水合现象,即一个Al原子与OH键合,同时与2个配位的水分子结合成为一个相对稳定的八面体[图9(B)][48].无定形SiO2颗粒表面具有孤立、连生和双生等3种类型的羟基[图9(C)~(E)][49].而C3N4表面主要由不完全缩合的氨基官能团与叔胺和芳胺组成[图9(F)].与SiO2载体相比,由于Al2O3载体表面碱性更强,因此在其催化剂表面上易形成稳定的Cu2Cl(OH)3[50][Scheme 1(Ⅰ),(Ⅱ)].PC-SiO2上活性金属与载体之间可以形成硅酸盐型配合物[Scheme 1(Ⅲ),(Ⅳ)][50].而PC-SiO2上存在的少量Cu2Cl(OH)3可能是由于CuCl2的水解而形成的.对于PC-C3N4而言,在含有大量CuCl2的制备条件中,CuCl2将与三-s-三嗪基序中的末端胺基反应,产生的副产物氯化氢(HCl)促使CN芳环打开[32],同时形成了无活性的Cu-N物种.因此,表面碱性较强的Al2O3载体更有利于Cu2Cl(OH)3活性相的形成,并使Pd,Cu物种之间的相互作用增强;而对于C3N4负载的Pd-Cu催化剂而言,其Cu物种更倾向与N原子配位而削弱了Pd,Cu之间的相互作用.可见,由于载体自身的特性使3种催化剂中形成不同的Cu活性物种,进而使Pd,Cu物种之间产生不相同的相互作用,最终导致不同载体负载的Pd-Cu催化剂的CO氧化活性性能不同.
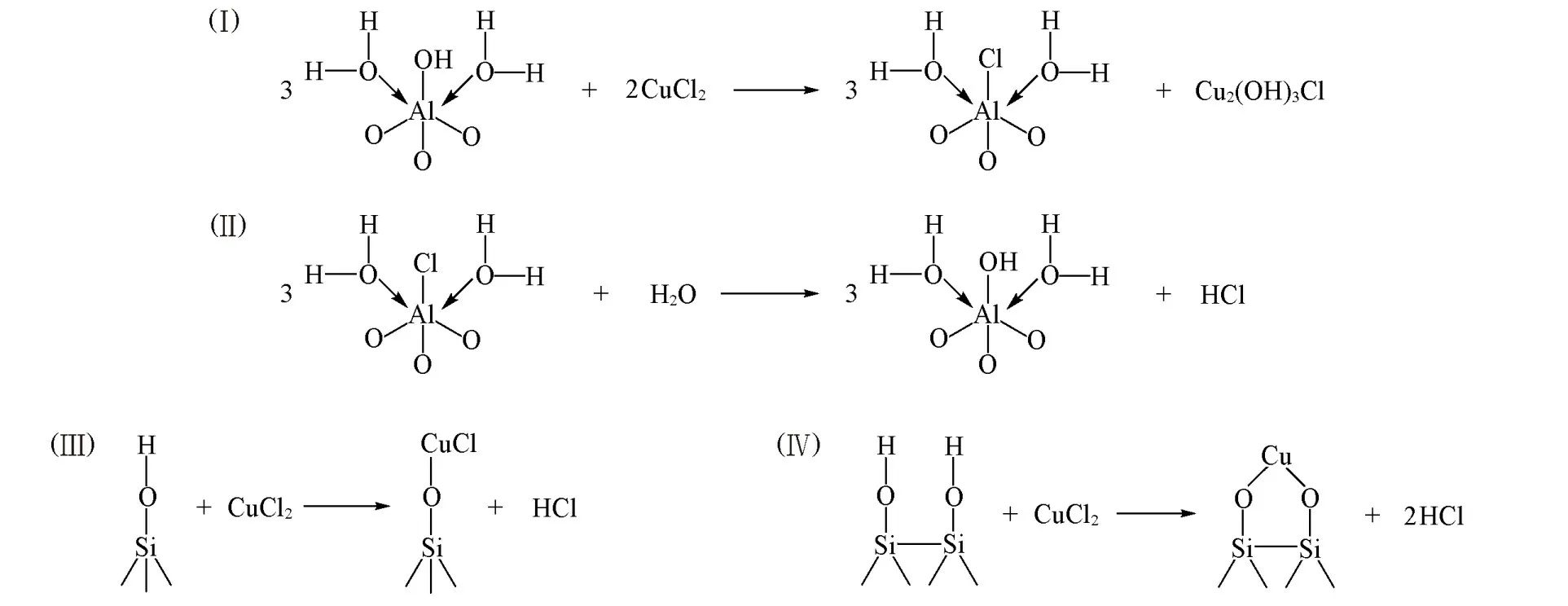
Scheme 1 Mechanism diagram of reaction between the active metal and the supports of three catalysts
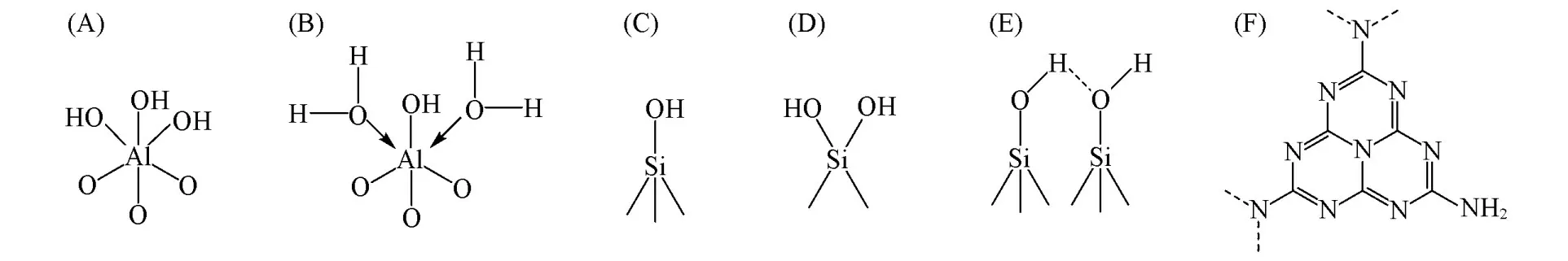
Fig.9 Surface hydroxyl types of the edge aluminum atoms(A,B)and amorphous SiO2(C—E)and the structure of g⁃C3N4(F)
以上3种负载型Pd-Cu催化剂在CO-PROX反应中表现出明显不同的催化活性,主要归因于载体的不同.就负载型Wacker催化剂而言,氧化态Pd物种和Cu2Cl(OH)3在其催化反应中起着至关重要的作用[21].在经典的Wacker循环中,吸附在活性Pd2+物种上的CO在水蒸汽分子的参与下很容易被氧化为CO2,同时Pd2+物种被还原为Pd0物种.此时相邻的Cu2(OH)3Cl或Cu(OH)Cl中的Cu2+将Pd0再氧化,并且Cu2+物种被还原为Cu+物种.最后,O2在Cu+上的吸附活化以促进Cu+到Cu2+物种的氧化.在上述循环中,Pd0的再氧化是CO氧化的第一决速步骤,同时O2氧化Cu+为Cu2+是另一速控步骤[43,44].在大量氢气存在时,Pd2+物种被CO还原为Pd0后,H2易吸附在Pd0物种上而导致其再氧化受阻,因此需要更多的氧化态Cu2+来氧化Pd0物种.而在实际的Wacker循环中,Cu2+的再形成源于O2对Cu+氧化,可见在催化循环中,Cu+的形成与转化同样至关重要.PC-Al2O3中存在较多的Cu2Cl(OH)3,并与Pd物种具有较强的相互作用,而PC-SiO2中Cu2(OH)3Cl较少且与Pd物种的相互作用较弱,导致其催化活性较低.PC-C3N4的Cu物种更倾向于与C3N4基体进行配位,与Pd物种的相互作用极弱,从而导致其无活性.进一步从XPS表征结果可知,与PC-SiO2相比,PC-Al2O3催化剂具有更多的表面Pd2+和Cu+物种.当通入反应气后,In situDRIFTs结果表明,PC-Al2O3中Pd2+迅速转变为更具活性的Pd+物种,甚至被还原为Pd0;同时引起Cu+物种活性中心发生变化,有利于实现Pd的再氧化,也再一次证实PC-Al2O3中Pd,Cu物种间具有较强的相互作用;而PC-SiO2在反应中仅有部分Pd2+被还原为Pd*和Pd0,Cu物种的吸附状态无明显变化,表明PC-SiO2中Pd,Cu物种间的相互作用较弱,同时Cu+物种含量远少于PC-Al2O3.对于PC-C3N4而言,一方面,表面Pd2+含量较低且对CO的吸附活化能力较弱;另一方面,其表面虽具有大量的Cu+物种,但其嵌入C3N4基质同样不利于CO的吸附,因此,PC-C3N4未表现出明显的催化活性.可见,与SiO2和C3N4相比,Al2O3更适合用于Pd-Cu催化剂的载体.基于Pd-Cu/Al2O3的结构性质,Scheme 2给出了该催化剂的表面结构示意图和可能的CO优先氧化反应机理.除经典的Wacker催化循环外,部分Pd2+物种转变为更具有活性的Pd+物种,Pd+物种吸附并氧化CO分子,自身被还原为Pd0物种.Cu2+物种将部分未被H2吸附的Pd0氧化为Pd+物种,同时Cu2+物种被还原为Cu+物种,最后,Cu+物种被吸附的O2氧化为Cu2+物种,从而实现催化循环.若Pd0被H2吸附后,将会抑制Pd0的再氧化,从而导致催化循环中断,这可能是CO转化率下降的原因.
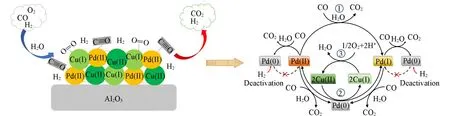
Scheme 2 Schematic mechanism of CO⁃PROX oxidation over Pd⁃Cu/Al2O3
3 结 论
以Al2O3,SiO2和C3N4为载体,通过简单浸渍法制备了PC-Al2O3,PC-SiO2和PC-C3N4等3种负载型Pd-Cu催化剂,并在连续流动微反应装置上考察了其富氢气氛下CO优先氧化性能.结果表明,相较于PC-SiO2和PC-C3N4两种催化剂,PC-Al2O3表现出较高的CO优先氧化活性.这可归因于Al2O3表面丰富的碱性位点有利于PC-Al2O3形成更多的Cu2(OH)3Cl物种,且与Pd物种产生更强的相互作用;在通入反应气后,PC-Al2O3表面易形成具有强CO活化能力的Pd+和更多Cu+物种,二者之间的强相互作用是维持催化剂活性的关键因素.与SiO2和C3N4相比,Al2O3更适合用于Pd-Cu催化剂的载体.该研究为探索新型CO优先氧化Pd基催化剂提供了参考.
